

 English
English文献解读|Signal Transduct Target Ther(52.7):皮质醇耐受性CAR-NK细胞可克服肺癌中类固醇诱导的免疫抑制
✦ +
+
论文ID
原名:Cortisol-resistant CAR-NK cells overcome steroid-induced immunosuppression in lung cancer
译名:皮质醇耐受性CAR-NK细胞可克服肺癌中类固醇诱导的免疫抑制
期刊:Signal Transduction and Targeted Therapy
影响因子:52.7
发表时间:2026.04.09
DOI号:10.1038/s41392-026-02638-z
背 景
肿瘤微环境(TME)通过建立免疫抑制微环境来逃避免疫监视。在TME中,缺氧、低pH值以及包括乳酸、氧化脂质和腺苷在内的免疫抑制代谢物的积累,会通过减少细胞因子产生和诱导耗竭表型来抑制细胞毒性T淋巴细胞(CTL)和自然杀伤(NK)细胞的效应功能。虽然类固醇的全身免疫抑制作用众所周知,但最近的研究表明,肿瘤可能利用从头合成类固醇作为逃避抗肿瘤免疫的另一种策略。特别是糖皮质激素已报道可促进转移。然而,局部糖皮质激素通过调节NK细胞功能来逃避抗肿瘤免疫的作用机制仍未得到充分研究。TME内免疫抑制性类固醇的积累可能源于局部类固醇生物合成、类固醇代谢、应激诱导的全身性升高,或作为癌症患者姑息治疗的一部分而使用的类固醇药物。与这种新兴观点一致,在多种实体恶性肿瘤中均已发现局部类固醇的产生或再生,包括结直肠癌、乳腺癌、胰腺导管腺癌和黑色素瘤,这提示肿瘤内糖皮质激素信号传导可能是实体瘤中一个尚未充分认识的免疫抑制通路。
实验设计
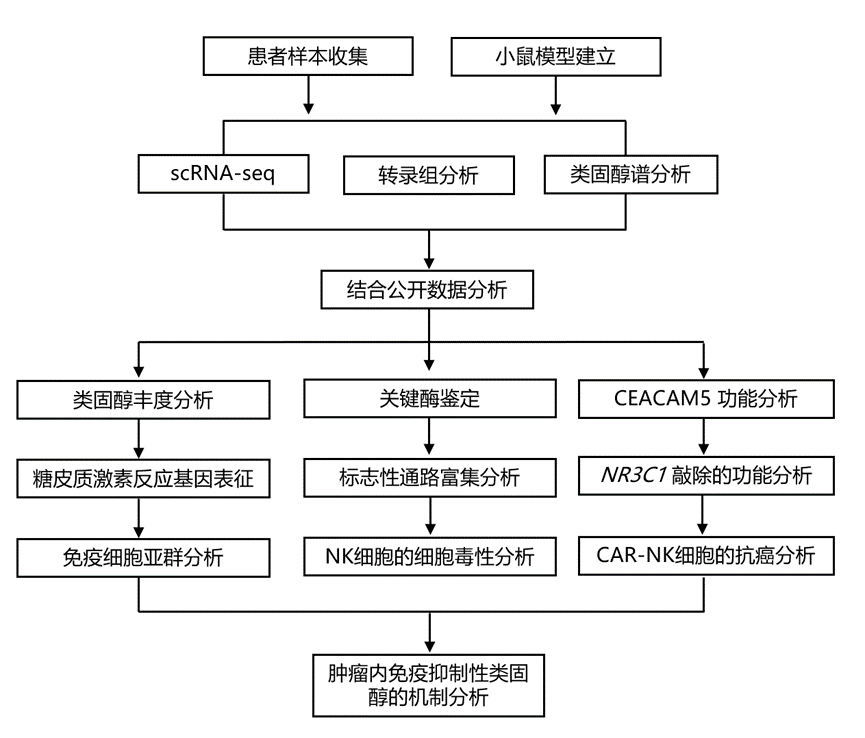
结 果
01

肺肿瘤微环境中的糖皮质激素信号传导抑制肿瘤浸润性NK细胞的功能
为了研究类固醇对肺肿瘤浸润性NK细胞的影响,研究团队首先定量分析了人肺TME中类固醇代谢物的丰度(图1a)。对34例患者的肿瘤样本进行靶向液相色谱-串联质谱(LC-MS/MS)分析显示,皮质醇是肺TME中最丰富的类固醇,平均浓度为42.47 ng/g肺肿瘤组织,其次是孕烯醇酮(24.7 ng/g)、可的松(9.7 ng/g)和皮质酮(4.38 ng/g)(图1b)。皮质醇(糖皮质激素)和孕烯醇酮(从头合成类固醇途径中所有类固醇的前体)是肺TME中富集的主要类固醇。对20例患者样本进行深入的类固醇谱分析,结果显示存在多种类固醇,包括糖皮质激素、盐皮质激素、雄激素、雌激素和孕激素。然而,未发现肺癌不同阶段皮质醇水平存在显著变化。
为了进一步研究糖皮质激素在肺肿瘤微环境中的功能效应,他们利用单细胞转录组分析(scRNA-seq)数据,分析了人NK细胞中受皮质醇调控的基因和通路,并比较了它们在肿瘤浸润NK细胞中的表达模式和丰度。对皮质醇处理的人原代NK细胞进行转录组学分析,鉴定出NK细胞中受糖皮质激素影响的基因特征,其中包括先前报道的糖皮质激素反应基因,例如TSC22D3、AREG、DAB2IP、FKBP5和IL7R(图1c)。在用氢化可的松(皮质醇)处理的活化人原代NK细胞中也观察到了类似的结果。他们观察到人原代NK细胞中几种皮质醇影响的激活标志物(NKG7、IFNG)和黏附标志物(ITGAL、ITGB7、SLAMF7、ICAM1)表达下调,而抑制标志物(KLRD1、CTLA4)表达上调。对7例非小细胞肺癌(NSCLC)患者的肿瘤和外周血样本进行scRNA-seq分析(GSE127465)显示,TME中皮质醇反应性转录程序显著增强(图1d-e)。值得注意的是,与低糖皮质激素受体(GR)表达的NK细胞相比,高GR表达的肿瘤浸润NK细胞表现出效应特征降低(NK细胞效应基因,包括PRF1、GZMB、IFNG、TNF、NKG7、KLRK1、DNAM1、EOMES、STAT5A和STAT5B的表达降低),表明类固醇介导的肺TME中NK细胞功能的抑制(图1f)。高GR表达的肿瘤浸润NK细胞表现出更高的功能障碍(NK细胞功能障碍基因,包括NR4A1、NR4A2、NR4A3、SOCS1、NFATC1和CBLB的表达升高)(图1g)。利用标志性通路进行深入的基因集富集分析 (GSEA) 表明,糖皮质激素不仅直接抑制 NK 细胞的功能,还能上调 NK 细胞中的缺氧基因特征(图1h)。scRNA-seq数据分析显示,在肿瘤浸润性 NK 细胞中,与GR低表达的NK 细胞相比,GR 高表达的 NK 细胞表现出更显著的缺氧应激特征(包括HIF1A、DDIT4、PFKFB3、ALDOA、CXCR4、ETS1、ATF3、JUN 和 HSPA5等缺氧诱导基因的表达)(图1i)。此外,在肺肿瘤NK细胞中,皮质醇反应评分与缺氧反应评分呈正相关(图1j)。
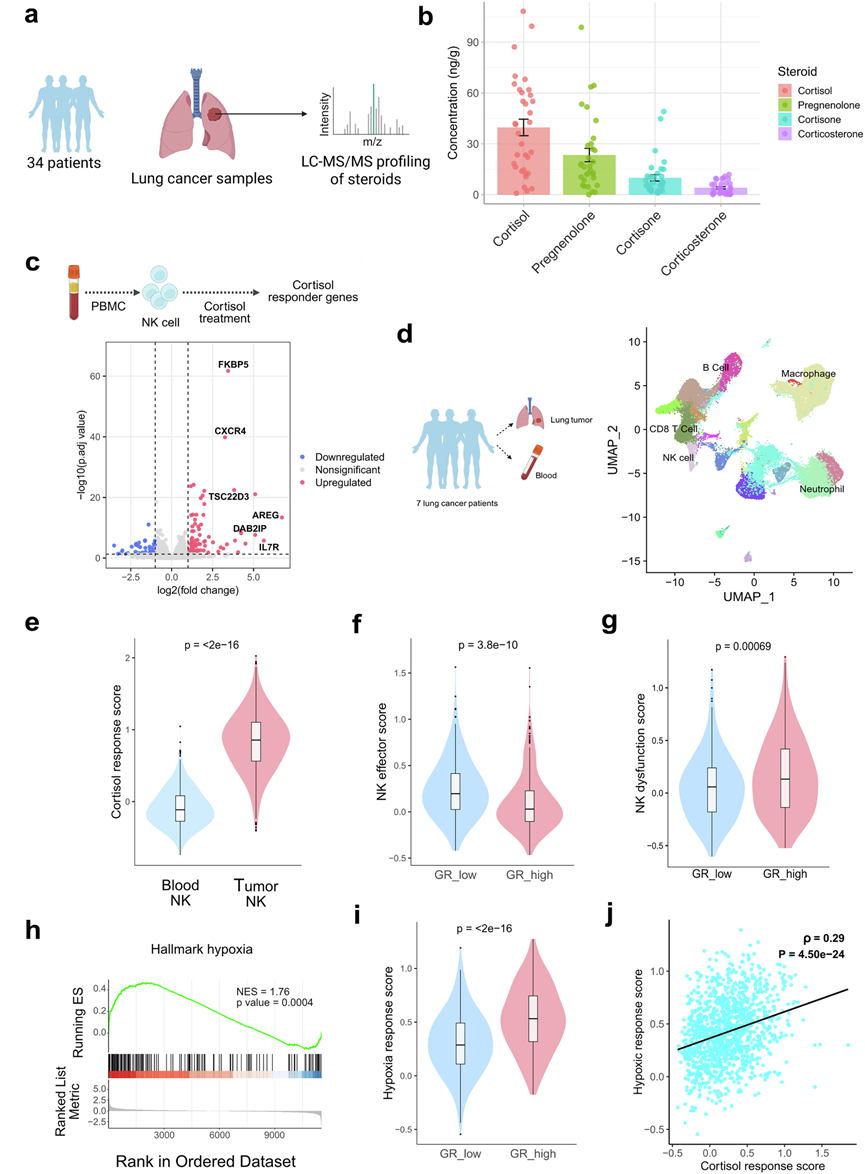
图1. 糖皮质激素对肺肿瘤浸润性NK细胞的影响。
(a) 使用LC-MS/MS对34例肺癌患者的类固醇谱进行分析的示意图。(b) 经有机溶剂萃取后,使用LC-MS/MS对34例患者样本中的类固醇进行定量检测。(c) 人原代NK细胞中皮质醇(一种糖皮质激素)反应基因的鉴定。(d) 七名患者(GSE127465)血液和肺肿瘤样本的scRNA-seq均匀流形近似和投影(UMAP)可视化图。(e) 小提琴图显示,肿瘤浸润NK细胞的皮质醇反应评分显著高于外周血NK细胞,表明肿瘤微环境中存在显著的糖皮质激素反应。(f) 小提琴图显示肺肿瘤微环境(TME)中糖皮质激素受体(GR)低表达与高表达NK细胞的效应功能评分,表明糖皮质激素反应会下调NK细胞的效应功能。(g) 小提琴图显示肺TME中GR低表达与高表达NK细胞的NK功能障碍评分,表明糖皮质激素反应会影响NK细胞功能障碍。(h) 基因集富集分析(GSEA)。(i) 肺肿瘤微环境中高糖皮质激素受体表达(GR-high)NK细胞的缺氧反应评分升高。(j) 散点图显示肺肿瘤浸润 NK 细胞中皮质醇反应评分与缺氧反应评分呈正相关。
02
CAF和巨噬细胞通过糖皮质激素代谢物的循环利用,使肿瘤内的皮质醇含量升高
鉴于肺TME中糖皮质激素含量丰富,且其对肺肿瘤浸润性NK细胞具有信号传导作用,他们试图鉴定导致肺TME中糖皮质激素富集的细胞。在从头合成类固醇的途径中,CYP11A1(也称为P450侧链裂解酶)负责将胆固醇转化为孕烯醇酮,其他下游类固醇由此合成(图2a)。在类固醇的代谢转化过程中,HSD11B1(也称为11β-羟类固醇脱氢酶1)负责将无活性的可的松转化为有活性的皮质醇。对来自不同数据集(GSE148071、KU_loom、GSE153935、GSE131907、GSE136246、GSE119911、GSE127465)的103例患者样本(包括非小细胞肺癌、鳞状细胞癌等不同分期(I期、II期、III期、IV期))的224611个细胞进行scRNA-seq,揭示了从头合成类固醇和糖皮质激素代谢物循环利用,并提高了肺TME中的皮质醇水平。T细胞、髓系细胞、成纤维细胞、巨噬细胞和癌细胞是类固醇从头合成的主要来源,且CYP11A1表达水平较高(图2 b-c)。相反,癌相关成纤维细胞(CAF)和巨噬细胞主要将无活性的可的松转化为有活性的皮质醇,同时HSD11B1表达升高(图2c)。在肺肿瘤微环境(TME)中,皮质醇生物合成基因的表达局限于免疫细胞和基质细胞(图2d)。虽然已知GR(由NR3C1编码)在所有活细胞中普遍表达,但T细胞、NK细胞和B细胞中GR表达升高,使其成为类固醇反应细胞(图2d)。然而,未观察到肺癌中HSD11B1和CYP11A1表达存在显著的阶段特异性差异(图2e)。在TCGA肺鳞癌(LUSC)队列中,HSD11B1高表达与较差的总生存期呈趋势性相关。
HSD11B1是一种能够将可的松再生为活性皮质醇的酶,主要在癌相关成纤维细胞(CAF)和肿瘤相关巨噬细胞(TAM)中表达。在髓系细胞中,HSD11B1的表达分布于各种巨噬细胞亚群,但在M2样TAM中显著富集(图2f)。对肺癌中表达HSD11B1的巨噬细胞进行GSEA显示,HSD11B1+ TAM的特征是异生物质代谢通路强烈激活,而p53、上皮间质转化(EMT)和TNFα-NF-κB信号通路则广泛抑制,这表明巨噬细胞处于代谢适应状态,炎症信号减弱(图2g)。对表达HSD11B1 的肿瘤相关巨噬细胞 (TAM)的转录谱分析进一步显示,免疫调节介质富集,包括CHI3L1、FBP1、LGALS2、CD38和强效 T 细胞抑制剂IL4I1,共同定义了具有明显免疫抑制潜力的皮质醇放大巨噬细胞群(图2h)。
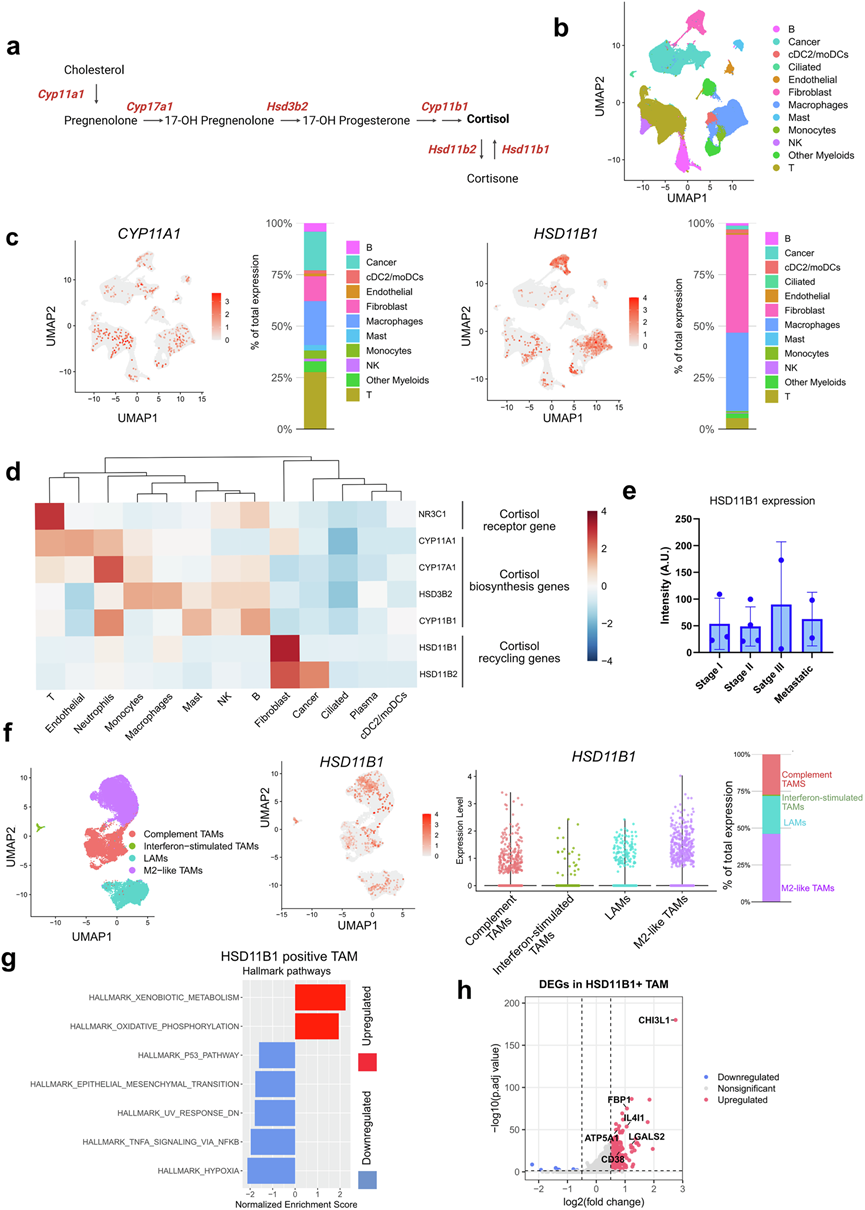
图2. 肺肿瘤微环境中糖皮质激素的从头合成和代谢循环。
(a) 示意图展示了从胆固醇到皮质醇的从头合成过程以及可的松代谢为皮质醇的过程。(b) 整合了包含 103 例肺癌患者的scRNA-seq数据集,展示了基于 UMAP 的主要细胞群聚类结果。(c) UMAP 特征图展示了所有细胞类型中CYP11A1(左)和HSD11B1 (右)的表达情况,以及相应的柱状图。(d) 热图总结了主要免疫细胞和基质细胞群中类固醇生成基因和皮质醇循环基因(的平均表达水平。(e) 免疫染色(共聚焦显微镜)分析,以检测HSD11B1的表达。(f) UMAP可视化和提琴图突出了主要亚群中巨噬细胞HSD11B1表达的异质性。(g) 通路富集分析。(h) 差异表达基因分析。
03
抑制糖皮质激素受体可增强肺肿瘤浸润性NK细胞的细胞毒性
在富含糖皮质激素的肺TME中,肿瘤浸润性NK细胞通过NR3C1编码的核受体GR对糖皮质激素产生反应。假设抑制该受体可能降低肺肿瘤NK细胞对糖皮质激素的反应性,并增强其细胞毒活性。为了验证这一假设,他们将LLC-OVA细胞系皮下注射到同系C57BL/6J小鼠体内,并使其形成肿瘤。实验组小鼠接受GR抑制剂米非司酮(60 mg/kg体重,隔日灌胃给药),对照组小鼠接受赋形剂(溶剂对照)。与赋形剂组相比,GR阻断组小鼠的肿瘤进展随时间推移显著减缓(图3a)。18天后,对肿瘤浸润NK细胞进行免疫表型分析显示,关键激活标志物(包括TNFα、IFNγ、穿孔素、DNAM1、NKG2D和CD69)表达上调,同时抑制性受体KLRG1表达下调,表明GR阻断小鼠体内NK细胞募集增强,且NK细胞具有更强的细胞毒性(图3b)。GR抑制还提高了表达TNFα和IFNγ的CD4+和CD8+T细胞的数量,与效应活性增强相一致。此外,GR阻断肿瘤中表达精氨酸酶(抗炎性M2型)的巨噬细胞数量减少,提示免疫微环境向促炎性方向转变。相反,脾脏NK细胞和CD4+/CD8+ T细胞的细胞毒性特征在GR阻断组和载体处理组之间保持不变,表明GR阻断重新激活了肿瘤驻留的糖皮质激素抑制的T细胞和NK细胞,而不是激活全身免疫。
肿瘤浸润NK细胞的比较转录组学分析显示,GR抑制组NK细胞与对照组存在显著差异(图3c)。在GSEA生物通路分析中,他们观察到GR抑制的肿瘤浸润NK细胞中急性炎症反应、2型免疫反应上调,促炎细胞因子(包括IL-1和IL-18)表达升高,以及ERK信号通路(与NK细胞活化相关)激活;而GR抑制则下调P38MAPK信号通路(应激激活通路),并对类固醇分解代谢过程产生负面影响(图3d-e)。他们还观察到GR抑制的肿瘤浸润NK细胞中PI3K信号通路显著上调,表明在糖皮质激素暴露下抑制的关键活化通路得以恢复(图3f)。对GR抑制小鼠和对照小鼠肿瘤浸润NK细胞的转录组进行比较分析,发现激活、细胞毒性、抑制和趋化性模块均存在显著的转录重编程。与这种激活模式一致,GR抑制也减弱了肿瘤浸润NK细胞中与缺氧相关的转录程序。关键缺氧调节因子的表达显著降低,表明糖皮质激素驱动的缺氧应激得到缓解。总而言之,这种转录转变表明,GR抑制可恢复NK细胞的激活和效应程序,同时减弱抑制信号,从而促进肺肿瘤微环境中NK细胞的细胞毒性表型。
在肺转移模型中,GR抑制显著降低了LLC-OVA细胞在肺部的定植(图3g)。GR抑制导致肿瘤浸润NK细胞活化增强,与对照组(载体处理小鼠)相比,这些NK细胞的IFNγ、CD107a和DNAM1表达均升高(图3h)。在肿瘤浸润的CD4+和CD8+T细胞中也观察到了类似的表型,GR阻断小鼠中产生IFNγ和TNFα的T细胞频率高于载体处理组。与皮下肿瘤模型的研究结果一致,脾脏NK细胞和T细胞的细胞毒性未发生显著变化,表明GR抑制选择性地恢复了肿瘤内免疫效应功能,而不影响全身免疫。这些结果提示糖皮质激素对肿瘤浸润NK细胞和T细胞的细胞毒性有负面影响,并加剧缺氧应激。阻断糖皮质激素信号传导可恢复NK细胞和T细胞的细胞毒性功能,并降低缺氧应激信号传导。
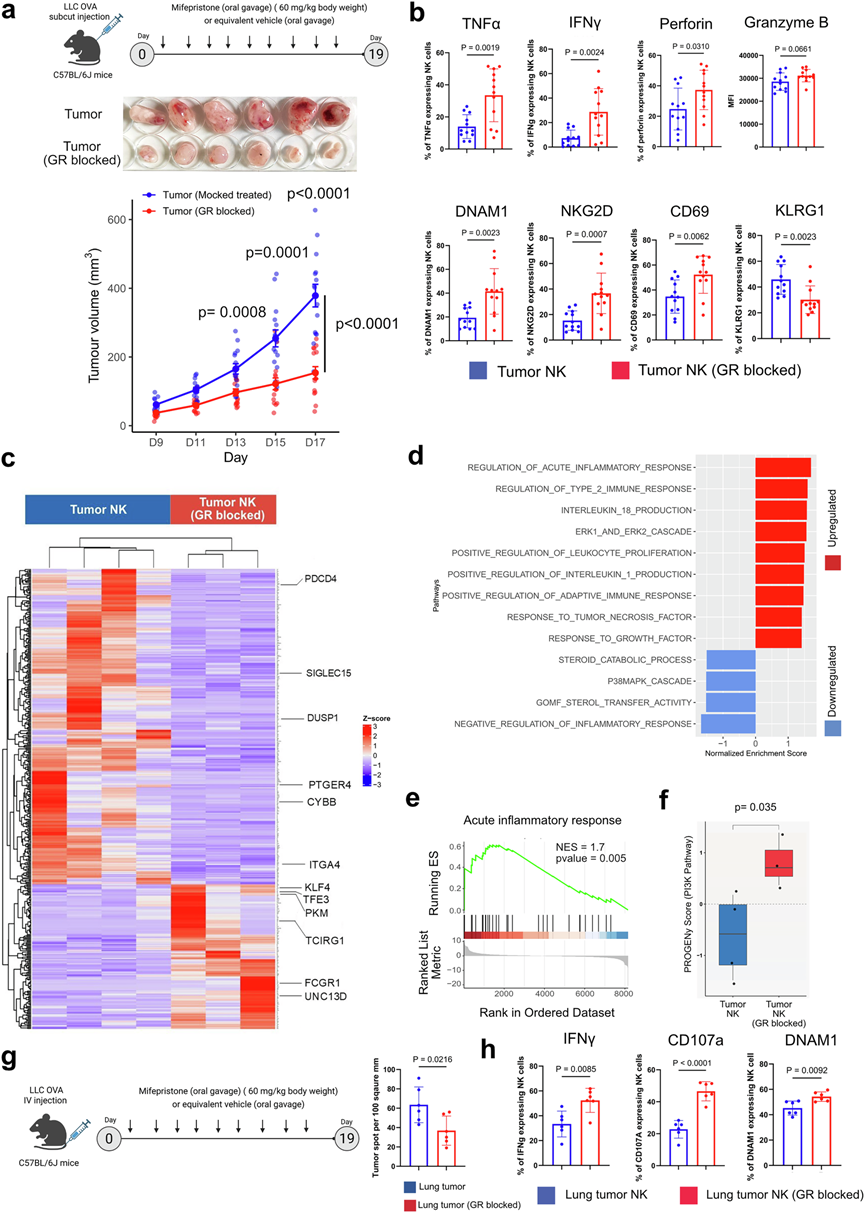
图3. 抑制糖皮质激素受体可增强肺肿瘤浸润性NK细胞的细胞毒性。
(a) 上图:在C57BL/6J小鼠中进行的研究示意图。中图:研究终点(第19天)肿瘤的代表性照片。下图:皮下肿瘤体积(mm3)随时间(天)变化的曲线图。(b) 流式细胞分析。(c) 热图显示了从GR阻断小鼠和载体对照组小鼠中分离的肿瘤浸润NK细胞的转录组谱。(d) GR阻断后肿瘤NK细胞中差异表达基因的GSEA。(e) GSEA富集图突出显示了GR阻断的肿瘤NK细胞中“急性炎症反应”标志性基因集的激活。(f) 来自PROGENy分析的通路活性评分。(g) 左图:静脉注射LLC-OVA肺转移模型的实验示意图。右图:肺肿瘤面积的代表性定量分析。(h) 流式细胞定量分析。
04
皮质醇耐受性CEACAM5特异性CAR-NK细胞在糖皮质激素存在的情况下也能杀死癌细胞
为了将糖皮质激素介导的免疫抑制概念应用于肺TME,他们旨在构建肺癌特异性糖皮质激素耐药的CAR-NK细胞用于免疫治疗。通过重新分析癌症基因组图谱(TCGA)数据集,他们发现CEACAM5(癌胚抗原相关细胞黏附分子5)在肺肿瘤中的表达显著高于正常肺组织,并通过促进细胞增殖和迁移驱动非小细胞肺癌的进展(图4a)。因此,他们对NK-92细胞系进行了基因工程改造,以构建CEACAM5特异性糖皮质激素耐药的CAR-NK细胞。使用了先前研究中报道的针对CEACAM5抗原的CAR构建序列。利用该序列,制备了微型质粒载体(GenCircle)。将编码 SB100X mRNA 的高活性 Sleeping Beauty 转座酶和编码 CEACAM-CAR 的 GenCircle 共转染至 NK-92 细胞系中,即可构建 CAR-NK 细胞(图4 b-c)。CAR 的表达随时间推移保持稳定。CEACAM5 特异性 CAR-NK 细胞与 A549 细胞系共培养 24 小时后,对过表达 CEACAM5 且 IFNγ 和 TNFα 表达升高的 A549 肺癌细胞系表现出显著的细胞毒性(图4d)。为了验证 CEACAM5 特异性 CAR-NK 细胞的特异性,他们选择了 CEACAM5 阴性的 RKO 细胞系,并构建了 CEACAM5 过表达的 RKO 细胞系(图4e)。CEACAM5特异性CAR-NK细胞能有效杀伤CEACAM5过表达的RKO细胞,但对CEACAM5阴性的RKO细胞系没有细胞毒性作用(图4e)。对与A549细胞共培养的CEACAM5特异性CAR-NK细胞进行转录组分析表明,CAR-NK细胞通过NF-κB信号通路激活,同时多种促炎细胞因子(包括IL-17、IL-12、IL-23、IL-1和IL-6)的产生相关基因表达富集。这种CAR特异性激活促进了NK细胞增殖并增强了黏附标志物的表达(图4f-h)。
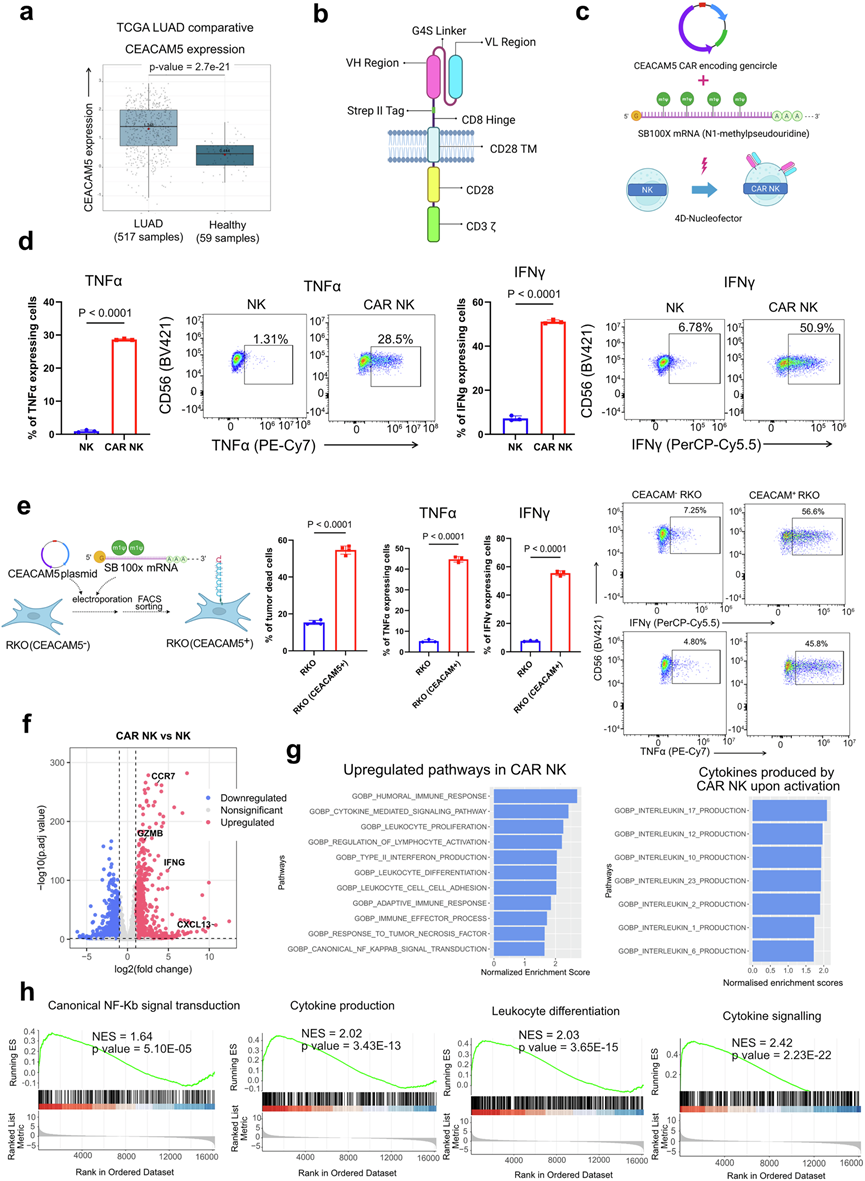
图4. CEACAM5特异性CAR-NK靶向表达CEACAM5的癌细胞系。
(a) TCGA肺腺癌样本和正常组织中CEACAM5表达上调。(b) CAR结构示意图。(c) 通过Sleeping Beauty技术构建CEACAM5 CAR-NK。(d) 流式细胞分析。(e) 由CEACAM5- RKO细胞系构建CEACAM5+ RKO细胞系。(f) 差异表达基因分析。(g) GSEA分析后,显示了显著上调的免疫通路。(h) 通过GSEA分析获得的活化CAR-NK细胞中主要上调的通路。
他们进一步通过敲除编码GR的NR3C1基因,构建了糖皮质激素耐药的CAR-NK细胞。利用CRISPR技术敲除NR3C1基因的第2外显子(编码N端结构域)和第5外显子(编码配体结合结构域),破坏了GR的结构及其配体结合能力(图5a)。为了筛选完全敲除NR3C1的CAR-NK细胞,对这些细胞进行了分选,并扩增了单个克隆。Sanger测序证实了所选克隆中NR3C1的敲除(图5a)。在皮质醇(氢化可的松,1 µM)存在的情况下,与传统CAR-NK细胞相比,皮质醇耐药的CAR-NK细胞(NR3C1敲除)对A549细胞系表现出更强的细胞毒活性(图5b)。这种增强的细胞毒性伴随着持续的杀伤活性以及IFNγ和TNFα表达的上调(图5c)。在皮质醇(氢化可的松,1 µM)存在下共培养48小时后,CAR-NK细胞的耗竭标志物PD-1显著升高,导致其杀伤活性受损。相反,糖皮质激素耐药的CAR-NK细胞未表现出耗竭迹象,并维持了其细胞毒活性(图5d-e)。对糖皮质激素耐药的CAR-NK细胞(NR3C1 -KO CAR-NK)和从富含皮质醇(1 µM)的A549共培养体系中获得的CAR-NK细胞进行转录组学研究,结果显示所有糖皮质激素反应基因(例如AREG、TSC22D3、FKBP5)均下调,这与最初在糖皮质激素耐药的CAR-NK细胞与CAR-NK细胞的比较实验结果一致。GSEA通路分析显示细胞毒性上调,并激活了多个通路,包括NF-κB炎症反应通路、IL-6/JAK-STAT3通路和IL-2/STAT5信号通路。然而,在CAR-NK细胞中,这些通路在皮质醇存在的情况下均下调。这些结果表明,糖皮质激素抑制CAR-NK细胞的功能,而NR3C1-KO CAR-NK细胞即使在糖皮质激素存在的情况下仍保持其杀伤活性。此外,转录组学研究显示,与CAR-NK细胞相比,皮质醇耐受的CAR-NK细胞中黏附标志物(ITGAX、ITGAM、SLAMF7、ITGAL、ICAM4)、活化标志物(CD244、CD226、LY9、IL2RB、CD69)表达上调,而抑制标志物(PDCD1、KLRD1、CTLA4、KLRC1)表达下调(图5f)。他们还发现趋化因子受体(CX3CR和CXADR)的表达升高。糖皮质激素耐药 CAR-NK 细胞中的多种趋化因子,表明与存在糖皮质激素的 CAR-NK 细胞相比,它在肺 TME 中具有更好的募集其他免疫细胞的潜力(图5f)。
为了阐明皮质醇诱导CAR-NK细胞功能障碍的机制,他们对暴露于皮质醇的活化CAR-NK细胞和NR3C1-KO CAR-NK细胞的转录组数据进行了PROGENy通路分析。值得注意的是,皮质醇暴露后,CAR-NK细胞中NK细胞活化和细胞毒性的两个关键调节因子PI3K-AKT和NF-κB信号通路显著受到抑制(图5g)。相反,NR3C1 -KO CAR-NK细胞在皮质醇处理下仍能维持(并有效恢复)PI3K-AKT和NF-κB信号通路的活性(图5g)。转录组分析结果一致表明,皮质醇处理的CAR-NK细胞中PI3K通路抑制基因选择性上调,而NR3C1敲除CAR-NK细胞中这些基因的表达基本保持不变,表明PI3K活性受到GR依赖性抑制。为了在蛋白水平上进一步验证这些发现,他们进行了免疫印迹分析,结果证实皮质醇处理的活化CAR-NK细胞中磷酸化AKT(p-AKT s473)和磷酸化IκBα(p-IκBα Ser 32/36)的水平降低,反映出PI3K-AKT和NF-κB通路的激活均受到抑制。值得注意的是,在皮质醇处理的活化NR3C1 -KO CAR-NK细胞中,这种抑制作用消失,表明GR信号的缺失在糖皮质激素应激下保留了这些关键的激活级联反应。综上所述,这些结果表明皮质醇通过抑制PI3K-AKT和NF-κB信号通路直接阻碍NK细胞的效应功能,而NR3C1的缺失则有效地恢复了这些通路,从而赋予了类固醇抗性。鉴于之前的观察结果表明皮质醇会加剧NK细胞的缺氧应激,接下来检测了活化CAR-NK细胞和NR3C1 -KO CAR-NK细胞在缺氧和缺氧+皮质醇联合条件下的转录组图谱。单独缺氧或缺氧联合皮质醇均能显著减弱CAR-NK细胞中NF-κB通路活性,其机制是通过降低磷酸化IκBα水平(图5h)。值得注意的是,NR3C1敲除的CAR-NK细胞在这些条件下仍保持了强大的NF-κB信号传导(图5h)。这种抵抗力表明,GR的基因敲除能够保护CAR-NK细胞免受缺氧诱导的信号传导崩溃的影响,使其即使在富含类固醇的肿瘤微环境中也能维持NF-κB介导的激活和细胞毒性。
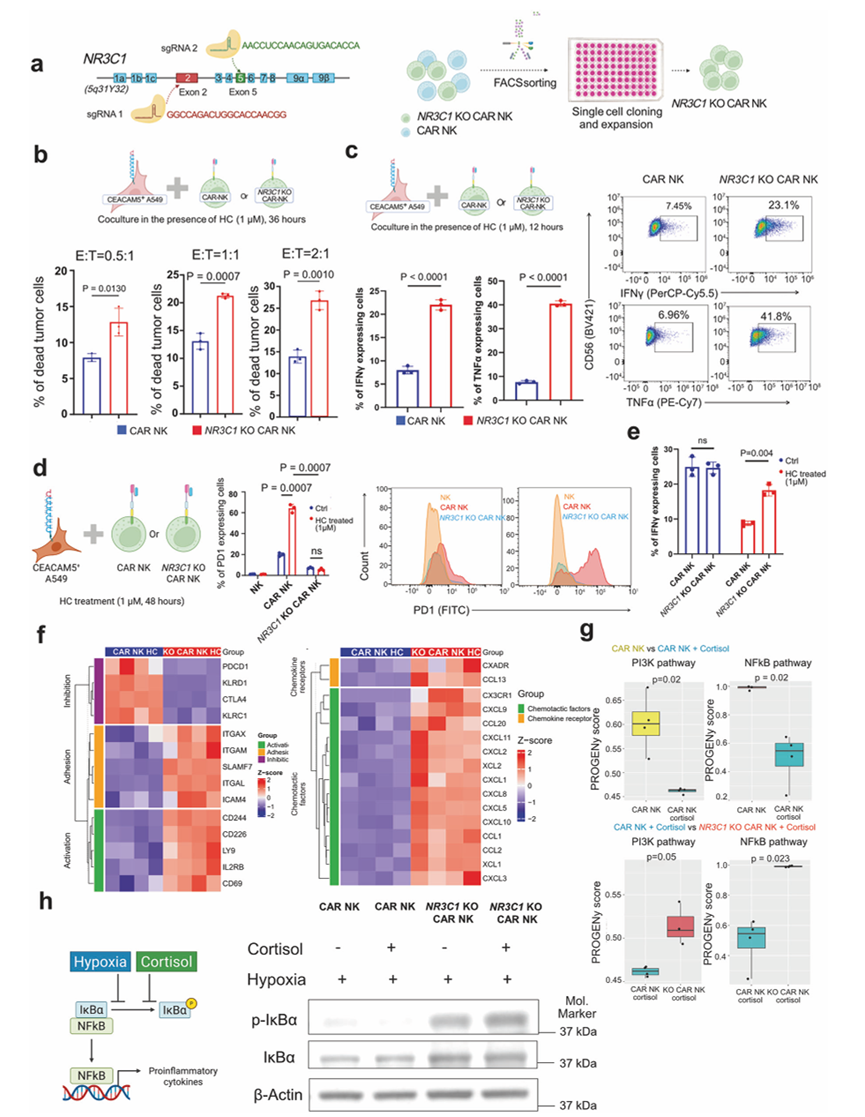
图5. 糖皮质激素抵抗ceacam5特异性CAR-NK细胞在糖皮质激素存在下消除肿瘤细胞。
(a) 糖皮质激素受体(NR3C1)基因的结构。(b) 在1 μM氢化可的松(HC,皮质醇)存在下,不同效应细胞与靶细胞(E:T)比例下, NR3C1 -KO CAR-NK细胞(皮质醇耐受型)和CAR-NK细胞与CEACAM5+ A549细胞共培养36小时后,对CEACAM5+ A549细胞的杀伤作用。(c) 基于流式细胞分析,绘制了 12 小时后表达 IFNγ 和 TNFα 的 CAR-NK 细胞和NR3C1 -KO 细胞的百分比。(d) 流式细胞分析测定PD1表达细胞的百分比。(e) 共培养48小时后,通过流式细胞分析获得IFNγ表达的CAR-NK细胞和NR3C1 -KO(皮质醇耐受性)CAR-NK细胞的百分比。(f) 热图展示了CAR-NK细胞和NR3C1 -KO CAR-NK细胞在与CEACAM5+ A549细胞共培养后,在氢化可的松(HC,皮质醇)存在下,关键功能基因模块的转录组谱。(g) PROGENy通路分析显示,皮质醇暴露后,活化的CAR-NK细胞中PI3K和NF-κB信号通路下调,而NR3C1-KO CAR-NK细胞在相同条件下均激活了这两个通路。
05
在富含类固醇的肺肿瘤模型中,皮质醇耐受性CAR-NK细胞的体内疗效
皮质醇耐受性(NR3C1 -KO)CAR-NK细胞优异的体外细胞毒性促使他们评估其体内抗肿瘤疗效。为了建立实验性肺转移模型,他们将1×106个萤火虫荧光素酶标记的A549细胞静脉注射到NSG SGM3小鼠体内( 每组n = 6)(图6a)。生物发光成像(BLI)证实14天后肺部已形成细胞定植。随后,将小鼠随机分为两组:一组在第15天接受1 × 107个常规CAR-NK细胞注射,另一组接受1×107个NR3C1 -KO CAR-NK细胞注射(图6a)。为了模拟富含类固醇的TME并促进NK细胞的持久性,两组小鼠均隔日接受地塞米松(10 mg/kg)和重组人IL-2(每只小鼠10000 U)联合治疗。纵向生物发光成像(BLI)监测显示,与CAR-NK细胞治疗组相比,NR3C1 -KO CAR-NK细胞治疗组的肿瘤生长显著减少(图6b-c)。组织病理学检查显示,皮质醇耐受的CAR-NK细胞治疗组的肿瘤负荷显著降低(图6d)。肺组织流式细胞术分析表明,NR3C1 -KO CAR-NK细胞维持了显著更高的颗粒酶B和CD107a表达,这与糖皮质激素暴露下持续的细胞毒性激活相一致(图6e)。虽然功能上存在优势,但两组小鼠肺部CAR-NK细胞和NR3C1-KO CAR-NK细胞的数量相当,它们在血液、骨髓、肝脏和脾脏中的分布也相似。CAR-NK组和NR3C1 -KO CAR-NK组小鼠的肝脏组织学表现也相似,两组小鼠的肝脏结构均保持完整,仅有轻微改变,包括肝细胞空泡化、肝窦和静脉淤血,以及极少的库普弗细胞活化或单核细胞浸润(图6f)。对 CAR-NK 和NR3C1 -KO CAR-NK 治疗的小鼠进行促炎细胞因子谱分析(IFNγ、IL-1β、IL-2、IL-4、IL-6、IL-8、IL-10、IL-13 和 TNFα),结果显示血清中存在低浓度(生理范围)的 IL-2 和 IL-8;然而,未能检测到其他促炎细胞因子(图6g)。
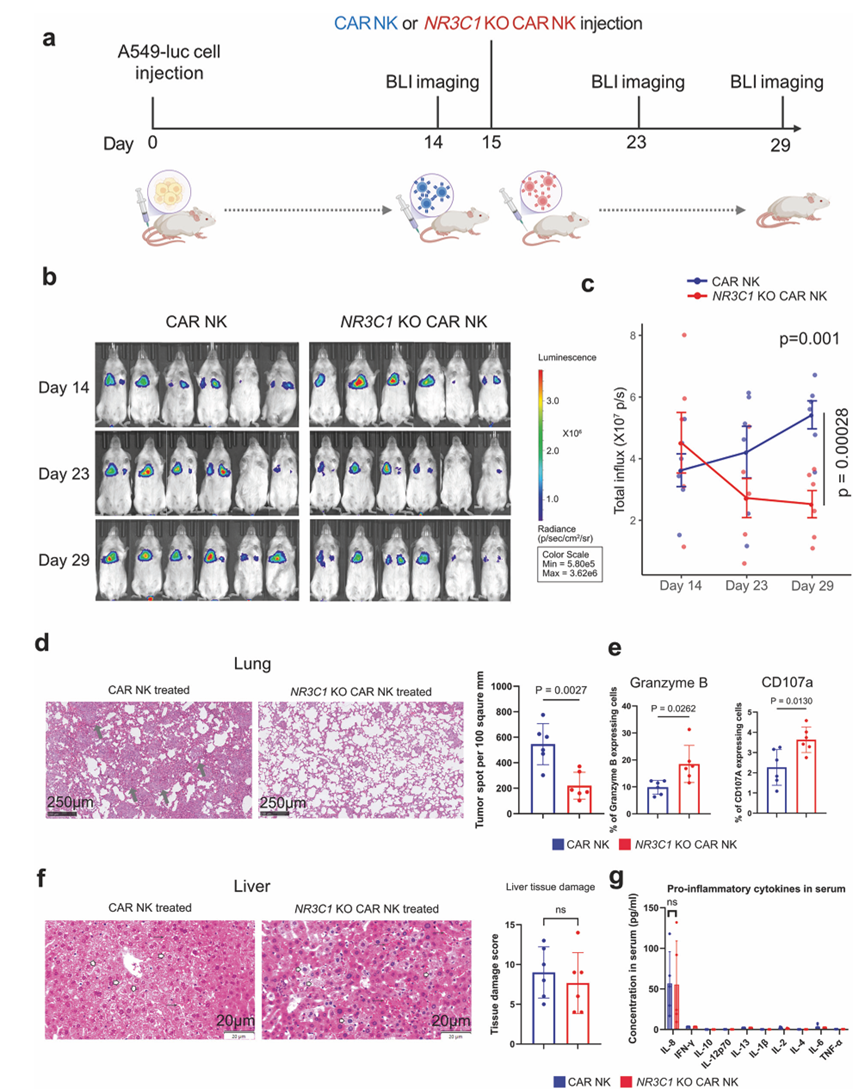
图6. 皮质醇耐受性CAR-NK细胞在体内表现出增强的抗肿瘤疗效。
(a) 实验流程示意图。(b) CAR -NK(左)或NR3C1 -KO CAR-NK(右)治疗小鼠的代表性连续BLI图像,显示NR3C1 -KO CAR-NK组在注射后14-29天内肿瘤逐渐消退。(c) 对肺部区域总生物发光通量随时间变化的定量分析显示,与 CAR-NK 对照组相比,NR3C1 -KO CAR-NK 细胞治疗的小鼠肿瘤负荷降低。(d) CAR-NK 和NR3C1 -KO CAR-NK 治疗小鼠的代表性H&E 染色肺组织切片。(e) 流式细胞术分析显示,与CAR-NK治疗的对照组相比,NR3C1 -KO CAR-NK治疗的小鼠肿瘤浸润NK细胞中CD107a+和颗粒酶B⁺细胞的频率更高。(f) CAR-NK(左)和NR3C1 -KO CAR-NK治疗组肝脏组织的代表性H&E染色显微照片。(g) CAR -NK 处理的小鼠和NR3C1 -KO CAR-NK 处理的小鼠在第 30 天血清中不同促炎细胞因子的浓度。
+ + + + + + + + + + +
结 论
本研究发现,肺TME中糖皮质激素皮质醇信号的功能性富集会削弱NK细胞的抗肿瘤细胞毒性并加剧缺氧应激。CAF和巨噬细胞可将无活性的可的松转化为有活性的皮质醇,而T细胞、成纤维细胞、髓系细胞、巨噬细胞和癌细胞则参与从头合成类固醇,共同构建了一个富含类固醇的微环境。体内GR的药理学抑制可缓解皮质醇介导的免疫抑制,从而减少肿瘤生长并增强肿瘤浸润性 NK 细胞的细胞毒性。为了克服皮质醇诱导的实体瘤靶向免疫疗法的功能障碍,构建了特异性靶向CEACAM5(在肺肿瘤中高表达)的CAR-NK 细胞,并通过基因敲除皮质醇受体基因NR3C1使其具有皮质醇耐受性。在富含皮质醇的微环境中,皮质醇耐受的 CAR-NK 细胞仍能维持抗肿瘤细胞毒性。机制上,NR3C1的缺失解除了皮质醇介导的 PI3K-AKT-NF-κB 信号通路的抑制,恢复了抗肿瘤活性,并显著降低了缺氧应激。在肺转移模型中,与传统CAR-NK细胞相比,皮质醇耐受性CAR-NK细胞实现了更优的肿瘤控制效果,并显著降低了肿瘤负荷。这些发现共同表明,局部皮质醇信号传导是实体瘤免疫治疗的关键障碍,并证实皮质醇耐受性CAR-NK细胞是一种靶向类固醇生成性实体瘤的有效策略,可与治疗性糖皮质激素联合使用。
+ + + + +



