

 English
English文献解读|Cancer Discov(33.3):整合蛋白质基因组学和正向遗传学揭示三阴性乳腺癌中一种新的有丝分裂脆弱性
✦ +
+
论文ID
原名:Integrative Proteogenomics and Forward Genetics Reveal a Novel Mitotic Vulnerability in Triple-Negative Breast Cancer
译名:整合蛋白质基因组学和正向遗传学揭示三阴性乳腺癌中一种新的有丝分裂脆弱性
期刊:Cancer Discovery
影响因子:33.3
发表时间:2025.11.03
DOI号:10.1158/2159-8290.CD-23-1173
背 景
在肿瘤发生过程中,癌细胞会获得癌基因和抑癌基因的改变,从而导致转录、代谢和生物合成过程的广泛变化。这些过程会促进不受控制的增殖和其他促肿瘤特征。然而,这些大分子合成和修饰的大规模变化也可能在癌细胞中产生新的依赖性和脆弱性。重要的是,这些相互抵消的促肿瘤和应激特征有时可能由相同的致癌突变引起。阐明癌症驱动基因如何赋予细胞应激以及癌细胞如何耐受这些应激表型已成为癌症治疗领域一个新兴的研究方向。三阴性乳腺癌 (TNBC) 是一种侵袭性乳腺癌亚型,目前有效的靶向治疗手段有限。紫杉烷类药物和其他微管靶向药物 (MTA) 是 TNBC 的一线化疗药物;然而,导致 TNBC 对紫杉烷类药物敏感的分子通路在很大程度上仍不清楚,这阻碍了对紫杉烷类药物敏感的患者筛选以及更具选择性的治疗策略的开发。
实验设计
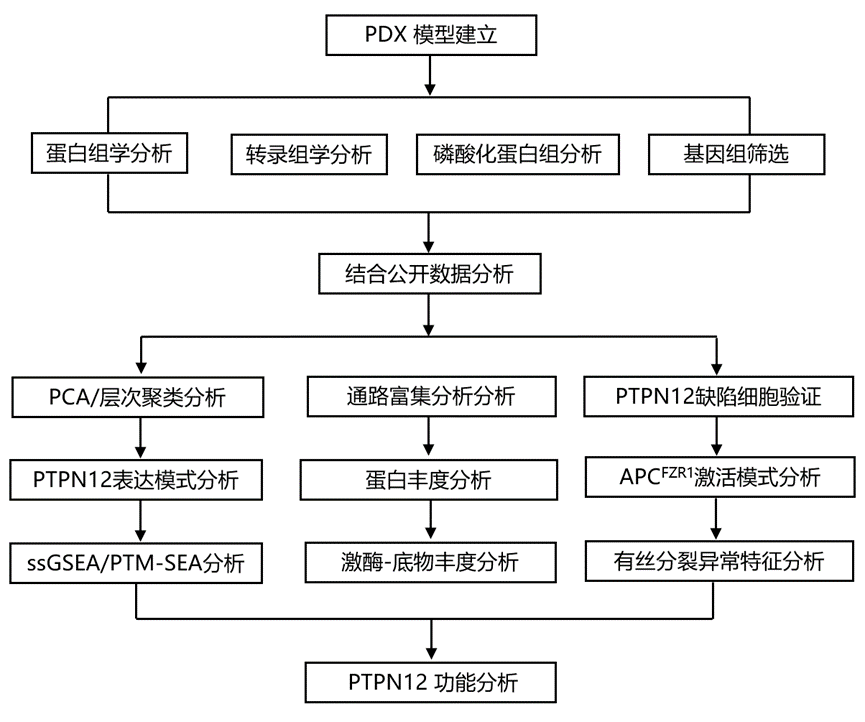
结 果
01

PTPN12缺陷型三阴性乳腺癌中蛋白质基因组学和功能遗传学的整合
由于肿瘤抑制因子PTPN12在TNBC中经常发生功能障碍,研究团队试图鉴定由PTPN12缺陷引起的分子改变和脆弱性。为此,他们整合了两种无偏倚的方法:对一组具有不同PTPN12状态的TNBC患者来源异种移植(PDX)模型进行大规模蛋白质组学表征,以及进行功能性遗传筛选以鉴定PTPN12合成致死通路(图1A-C)。蛋白质组学已广泛用于鉴定特定疾病状态(包括癌症进展过程中)中发生改变的分子通路。然而,这种方法的局限性在于,仅基于肿瘤样本的相关性分析,很难识别特异性驱动癌症进展的改变,而非疾病状态的继发效应。因此,为了鉴定对 PTPN12 缺陷细胞生长也有功能性影响的失调通路,他们将从 PTPN12 缺陷 TNBC 模型的蛋白质组学表征中获得的信息与基因筛选相结合,以识别由 PTPN12 失活产生的选择性脆弱性。
他们对一组源自14例独立TNBC患者的PDX模型进行了蛋白质组学分析,这些模型中PTPN12蛋白的表达水平和功能存在异质性。从每个PDX模型中获取三个独立的肿瘤样本,并采用全外显子组测序、转录组分析(RNA-seq)以及蛋白质和磷酸化位点的定量蛋白质组学方法进行分析(图S1A)。初步筛选后,获得了超过19000个基因和10000个不同蛋白质的拷贝数、转录本和肽段丰度数据。此外,他们还鉴定了约40000个磷酸化位点,这些位点来源于超过7000个磷酸化蛋白。在所有分析的数据类型中,生物学重复样本均高度相关,表明数据集内模型内部差异较小,模型间异质性较高,mRNA 和蛋白质之间的相关性中位数为 0.55,与先前对原发性乳腺肿瘤的蛋白质组学研究结果一致(图 S1B-C)。重要的是,使用无监督分层聚类将本研究的PDX 队列与原发性乳腺癌样本进行比较,结果表明,这些模型的基因组和转录谱能够忠实地代表基于 PAM50 分类的基底细胞亚型(图 S1D-F)。
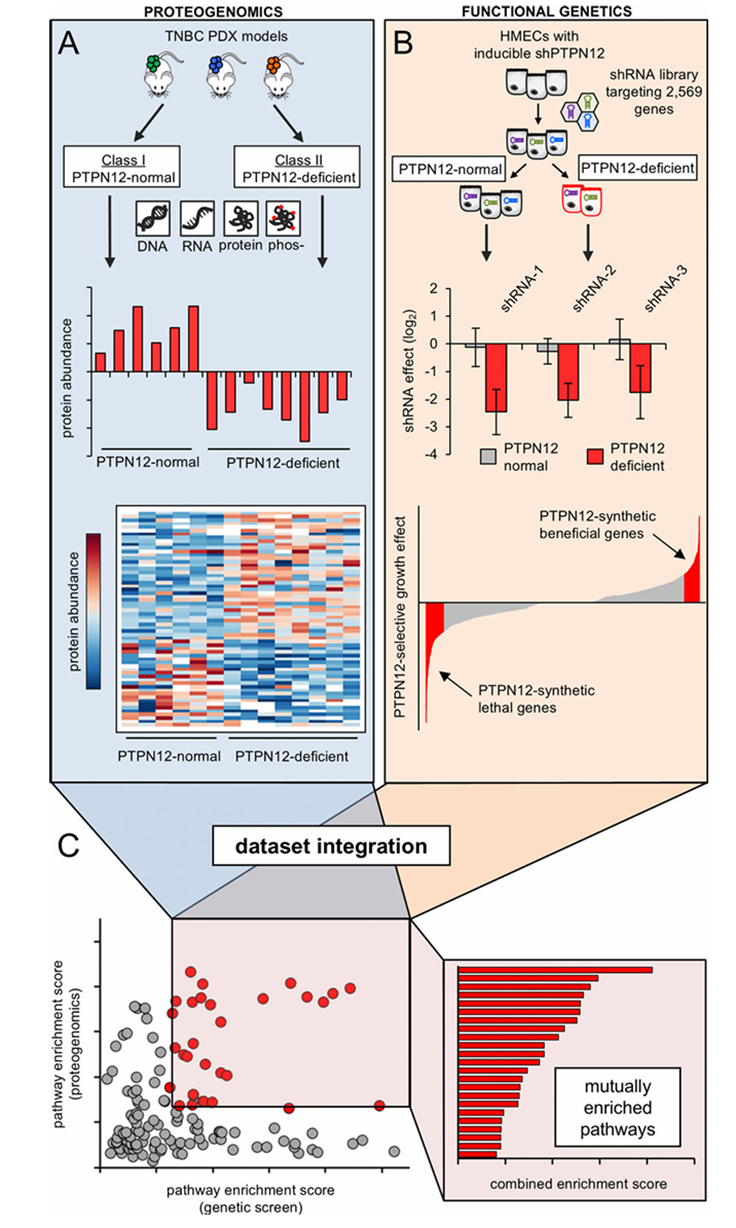
图1. 整合蛋白质基因组学和正向遗传学以揭示 TNBC 的新靶点。
(A) 对一组TNBC患者来源异种移植(PDX)模型进行了蛋白质组学表征。(B) 基因筛选,以识别在PTPN12缺陷细胞中选择性必需的基因。(C) 利用基因集富集分析(GSEA)整合。
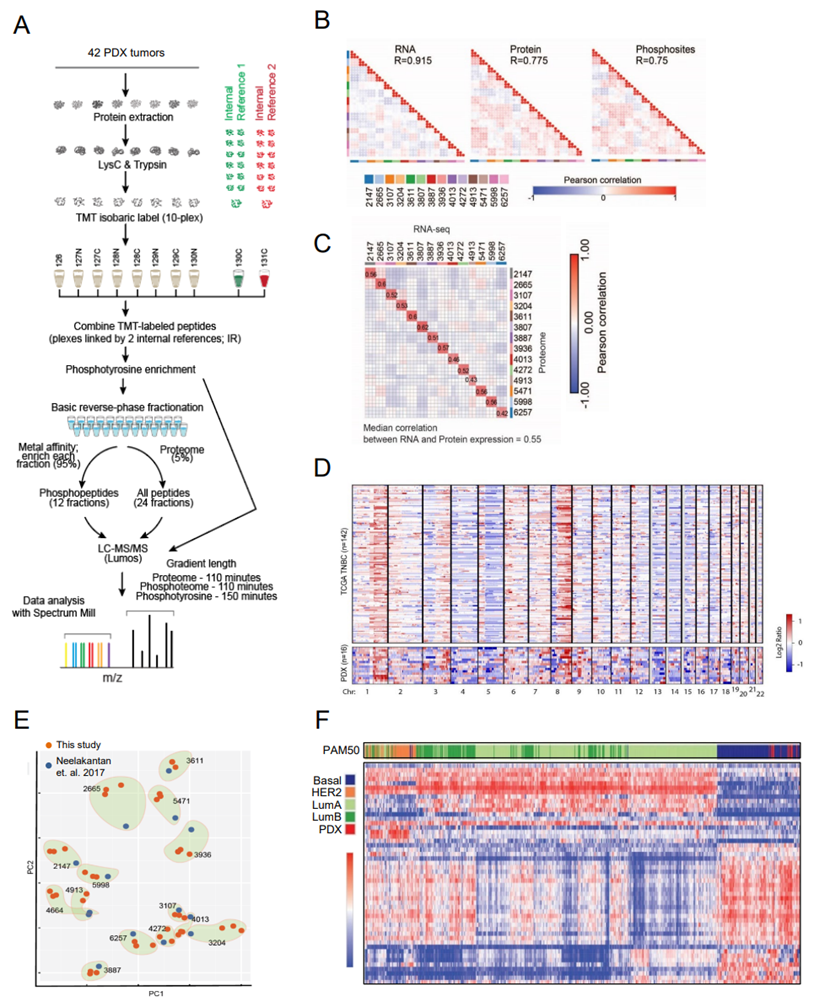
图S1. 结合蛋白质基因组学和功能遗传学揭示TNBC的新脆弱性。
(A) 蛋白质组学分析的示意图。(B) 热图显示了在RNA、蛋白质和磷酸化蛋白质组中所有三份PDX模型之间的Pearson相关性。(C) 热图显示mRNA和蛋白质表达之间的样本Pearson相关性。(D) PDX样本与TCGA三阴性乳腺肿瘤拷贝数畸变谱的比较。(E) 将本研究中 PDX 模型的转录组谱与先前使用相同模型的研究进行主成分分析比较。(F) PDX 转录组数据与 TCGA 原发性乳腺癌样本的层次聚类中,显著下调的肽段在左侧有标注。
02
PTPN12缺陷型肿瘤中RTK信号传导和细胞周期进程通路失调
他们首先根据 PTPN12 的表达水平将 PDX 模型分为两组,以便识别 PTPN12 低表达和高表达模型之间的差异表达特征。由于既往研究表明,PTPN12 蛋白水平适度降低(40-50%)足以驱动异常信号传导和细胞转化,因此他们根据免疫组化 (IHC) 检测的正常乳腺上皮样本中 PTPN12 的表达水平来定义 PTPN12 状态(正常或缺失)。重要的是,他们观察到蛋白质组学和 IHC 检测的 PTPN12 表达水平具有良好的相关性。基于此分类,他们确定了 PTPN12 正常模型和 PTPN12 缺失模型之间每个基因、蛋白和磷酸化位点的差异表达。对这些数据集进行的单样本基因集富集分析(ssGSEA)和 PTM-SEA分析揭示了 PTPN12 缺陷模型中两大类通路失调(图 2A)。首先,他们发现与生长因子受体信号通路相关的通路在磷酸化蛋白质组水平上显著上调。这与先前观察到的结果一致,即 PTPN12 缺陷会导致 TNBC 中一组致癌受体酪氨酸激酶的协同激活。在失调最显著的激酶中,他们观察到 MET、PDGFR 和 HER 家族 RTK 的磷酸化和活性增强,这些激酶此前已证实会在 PTPN12 缺陷肿瘤中过度磷酸化(图 2B-C)。其次,他们发现PTPN12缺陷模型中参与细胞周期和有丝分裂进程调控的通路在转录组、蛋白质组和磷酸化蛋白质组水平上均出现显著下调(图2A)。重要的是,在PTPN12缺陷模型中,调控有丝分裂进程和检查点信号传导关键环节的激酶(例如CDK1、CDK2、PLK1、AURKB)的表达及其下游靶标的磷酸化水平均降低,这表明PTPN12缺陷模型可能因此出现这些过程的缺陷(图2B-E)。通过基于UniProt关键词“mitosis”的蛋白质表达进行无监督聚类分析,进一步阐明了PTPN12缺陷模型中独特的有丝分裂蛋白表达谱。结果表明,PTPN12缺陷模型的有丝分裂蛋白表达谱高度相似,与PTPN12正常模型的表达模式截然不同。这些发现突出了 PTPN12 缺陷型 TNBC 模型中失调的两条生物学途径,并代表了 TNBC 的这一亚群的潜在依赖性:生长因子信号传导和有丝分裂调节。
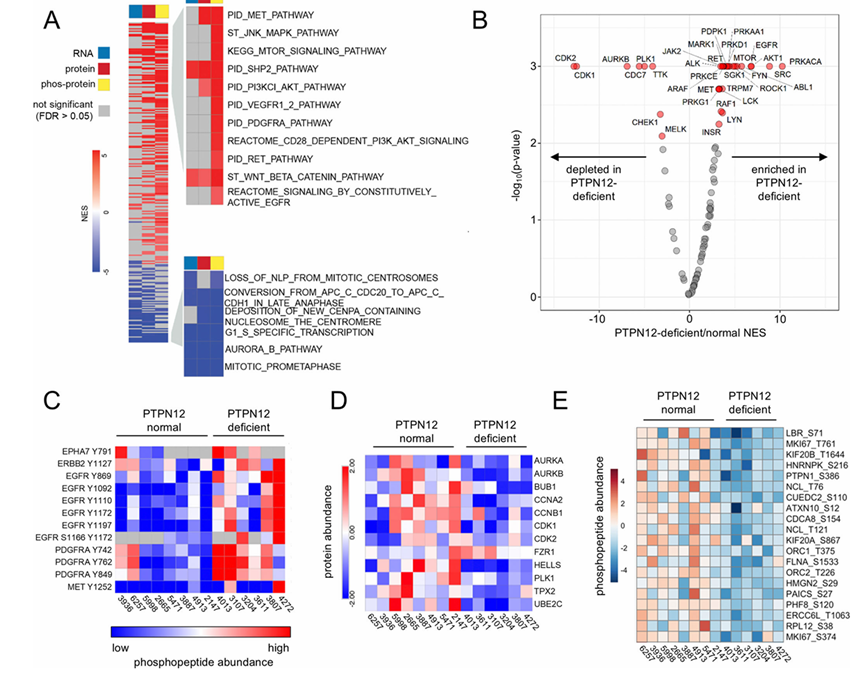
图2. PTPN12 缺陷模型表现出细胞周期和有丝分裂途径的下调。
(A) 对 PTPN12 正常模型和缺陷模型进行修正双样本 t 检验,并转换 p 值后进行 ssGSEA 分析。(B) 基于磷酸化蛋白质组学的 PTM-SEA 分析显示,PTPN12 调控的酪氨酸激酶特征(例如 EGFR、MET、ABL1)显著上调,而有丝分裂激酶特征(包括 PLK1、CDK1、CDK2 和 AURKB)显著下调。(C) 在PTPN12缺陷的PDX模型中,PTPN12调控的RTK中的磷酸酪氨酸位点过度磷酸化。(D) 在PTPN12缺陷的PDX模型中,参与细胞周期进程和有丝分裂的蛋白质表达下调。(E) PTPN12缺陷型和PTPN12正常型模型中细胞周期和有丝分裂相关蛋白的标准化蛋白丰度热图。(F) PTM-SEA富集的20个细胞周期和有丝分裂相关激酶(PLK1、CDK1、CDK2或AURKB)的磷酸化肽底物的标准化丰度热图。
03
PTPN12缺陷细胞对有丝分裂调节因子的进一步扰动很敏感
为了补充蛋白质组学分析,他们试图鉴定与PTPN12缺陷产生合成致死效应的蛋白质和通路,并评估这些通路作为潜在治疗靶点的价值。为此,他们进行了正向遗传筛选,以鉴定在永生化人乳腺上皮细胞(shPTPN12-HMEC)中具有PTPN12选择性生长效应的基因。这些永生化人乳腺上皮细胞经强力霉素诱导的短发夹RNA(shRNA)基因工程改造,该shRNA靶向PTPN12(图1B)。合成致死筛选是通过将shPTPN12-HMEC转导逆转录病毒shRNA文库而进行的,该文库包含24874个shRNA,靶向2569个基因(约10个shRNA/基因),其中包括激酶、磷酸酶、泛素连接酶和染色质修饰酶等具有成药潜力的酶。将转导细胞在有或无PTPN12-shRNA诱导的情况下进行培养。分别在两个时间点(嘌呤霉素筛选后立即和培养18天后)提取基因组DNA,并通过二代测序分析扩增的shRNA序列。比较载体组和强力霉素处理组中每种shRNA丰度随时间的变化,以鉴定对细胞生长和存活具有PTPN12选择性作用的shRNA。
通过合成致死筛选和蛋白质组学分析筛选出的基因和通路整合起来,以鉴定在PTPN12缺陷型TNBC模型中特异性失调的靶点,这些靶点在PTPN12缺陷细胞中受到干扰时会导致选择性生长抑制。合成致死筛选鉴定出102个基因,这些基因在PTPN12缺陷型HMEC细胞中选择性地表现出显著的生长效应(图3A)。有趣的是,基于蛋白质-蛋白质相互作用和文本挖掘的这些候选基因的网络分析揭示了一个参与有丝分裂调控和检查点信号传导的基因聚类,包括PLK1、BUB1和后期促进复合物(APC)的几个组分,这些基因在PTPN12缺陷型PDX模型中也表现出蛋白水平的异常表达(图3B-C)。这一发现表明,PTPN12缺陷细胞不仅表现出细胞周期和有丝分裂相关蛋白表达失调,而且对调控这些过程的特定蛋白的进一步扰动也十分敏感。与此假设一致,对蛋白质组学和遗传筛选的基因集富集分析进行直接比较发现,两个数据集中均富集的通路通常属于三个功能类别:有丝分裂进程和纺锤体检查点的调控、APC底物的调控以及G1/S期进程的调控(图3D-E)。
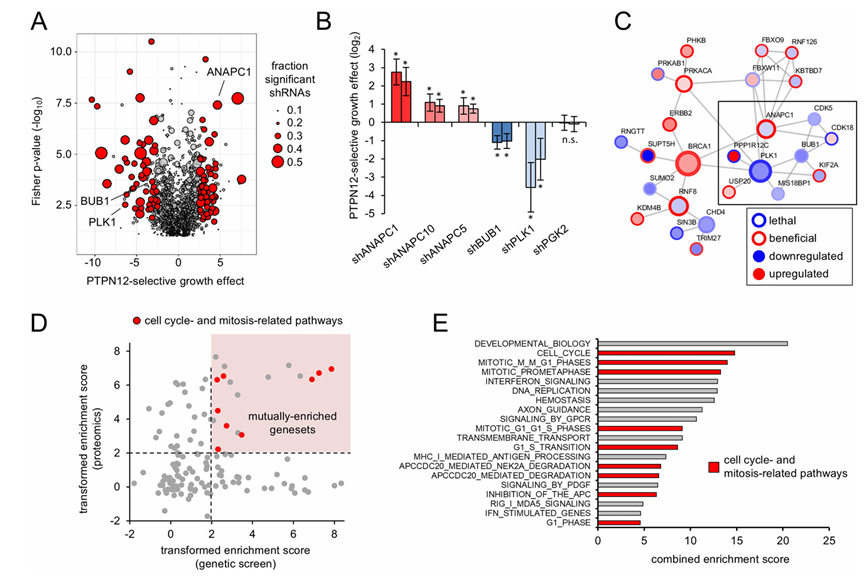
图3. PTPN12 缺陷细胞容易受到有丝分裂调节因子的扰动。
(A) 火山图展示了合成致死筛选的结果,用于鉴定PTPN12选择性生长调节因子。(B) PLK1和BUB1的敲低导致PTPN12合成致死,而多个APC组分的敲低则导致PTPN12合成有益。(C) shRNA筛选和蛋白质组学显著结果的网络分析。(D) 蛋白质组学和基因筛选数据集的GSEA分析。(E) 与细胞周期进程调控和有丝分裂检查点信号传导相关的REACTOME通路在基因筛选和蛋白质组学数据集中均富集。
04
PTPN12 缺乏导致 APCFZR1过度激活
基于蛋白质组学数据中APC相关通路富集以及敲低多种不同APC亚基可选择性地挽救PTPN12缺陷细胞生长缺陷的发现,他们假设PTPN12失活可能导致APC活性失调。为了验证这一假设,他们首先评估了PTPN12免疫组化状态与TNBC PDX模型队列中APC底物丰度之间的关联,这些模型已进行了蛋白质组学表征。该分析显示,PTPN12缺陷模型中许多APC底物的丰度明显降低,提示APC在PTPN12缺陷肿瘤中可能过度激活(图4A)。他们还在77例TCGA乳腺癌样本中验证了这种关联,发现原发性人乳腺肿瘤中APC底物丰度也与PTPN12水平呈正相关。APC是细胞周期进程的关键调控因子,在有丝分裂中期/后期和G1/S期转换中均发挥作用,其活性分别受两个主要共激活因子CDC20和FZR1的调控。因此,为了区分不同形式的APC复合物的失调,他们检测了PTPN12蛋白水平与这两个共激活因子蛋白磷酸化水平之间的相关性。有趣的是,他们发现PTPN12丰度与FZR1磷酸化水平呈显著正相关,但与CDC20磷酸化水平无相关性,这表明FZR1失活磷酸化的缺失可能导致PTPN12缺陷型肿瘤中APC FZR1的特异性过度激活(图4B)。与此假设以及 APC FZR1在抑制 G1/S 转换中的作用一致,PTPN12 缺陷的 PDX 模型在体内表现出明显较慢的生长速度。
为了探究FZR1低磷酸化及其导致的APC FZR1过度激活的潜在机制,他们重新分析了从TNBC PDX模型中生成的蛋白质组学数据,以寻找PTPN12缺陷模型中失调的激酶。通过基于磷酸化蛋白质组学数据中底物磷酸化水平推断激酶活性,观察到CDK2是PTPN12缺陷PDX模型中下调最显著的激酶之一(图2B)。鉴于CDK2通过磷酸化FZR1抑制APC FZR1活性的关键作用,这些结果提示CDK2的抑制可能是APC FZR1过度激活的潜在机制。为了验证这一假设,他们发现敲低 CDC25A 或 CDC25B(两种部分非冗余的磷酸酶,它们通过去磷酸化保守的 Y15 残基 30-32 来激活 CDK2)与PTPN12失活具有合成致死性(图 4C)。相反,敲低 WEE1(一种磷酸化 CDK2 Y15的酶)则选择性地促进了 PTPN12 缺陷细胞的生长(图 4C)。综上所述,这些结果表明 PTPN12 失活导致 CDK2 活性受到抑制,这可能是通过 CDK2 Y15的过度磷酸化实现的,从而损害了 PTPN12 缺陷细胞的增殖。为了验证该模型,他们构建了带有 CDK2 活性报告基因的 HMEC 系统,发现敲低 PTPN12 会显著抑制 G1 期 CDK2 的激活(图 4D)。他们还进行了双分子荧光互补 (BiFC) 实验,以评估 PTPN12 是否与 CDK2 相互作用,结果发现二者之间存在强烈的相互作用,类似于 EGFR 和 PTK2B(PTPN12 的两个已知相互作用蛋白)的相互作用。此外,他们观察到敲低 PTPN12 会增加 CDK2 Y15 的磷酸化水平,这支持了 PTPN12 失活导致 CDK2 Y15过度磷酸化从而抑制 CDK2 的观点,进而阻碍G1/S 期转换过程中APCFZR1的正常失活。此外,这些发现表明,异常的 APCFZR1活性可能导致下游 APCFZR1底物的耗竭,而这些底物对于细胞周期和有丝分裂进程至关重要。
为了验证这一预测,他们首先构建了带有FUCCI报告基因系统的HMEC细胞,并在转染对照siRNA或靶向PTPN12的siRNA 35 72小时后进行流式细胞分析。有趣的是,PTPN12敲低导致大量细胞在G1期晚期(CDT1+/GMNN−)和S期早期(CDT1+/GMNN+)积累,这与APC FZR1过度激活导致的G1/S期进程延迟相一致(图4E)。接下来,他们利用同一系统,通过siRNA介导的PTPN12敲低,并利用活细胞成像技术监测AmCyan-GMNN的丰度,来检测PTPN12缺失是否会导致S期GMNN(APCFZR1的特异性底物)的正常积累缺陷。事实上,他们观察到PTPN12缺陷细胞中AmCyan-GMNN的积累速度比对照细胞慢,这表明PTPN12缺陷细胞中G1/S期转换过程中APC FZR1的正常失活受到干扰(图4F)。重要的是,他们发现用WEE1抑制剂adavosertib治疗可以挽救这种表型,这支持了CDK2 Y15过度磷酸化驱动PTPN12缺陷细胞中APC FZR1异常激活和G1/S期进程受损的模型(图4G)。
基于这些结果,他们假设PTPN12缺陷细胞中APC FZR1的异常活性也可能导致G2期和有丝分裂期间APCFZR1底物的异常降解。为了验证这一假设,他们对shPTPN12-HMEC细胞进行了免疫荧光染色,以检测pH3+细胞在G2期晚期和M期早期中几种经典APCFZR1底物(PLK1、AURKB和CCNB1)的丰度。PTPN12敲低导致pH3+细胞中所有三种APC FZR1底物的异常耗竭,这与PTPN12缺陷细胞中APCFZR1在G1/S期转换后仍保持异常活性的假设相符。重要的是,在PTPN12缺陷的pH3+细胞中,PLK1丰度的降低可以通过APC抑制剂proTAME来恢复,这直接表明APC过度激活与该表型有关(图4H-I)。综上所述,这些结果支持以下假设:PTPN12的扰动会导致APCFZR1的异常激活,并在细胞周期的多个阶段导致APCFZR1底物的异常降解。
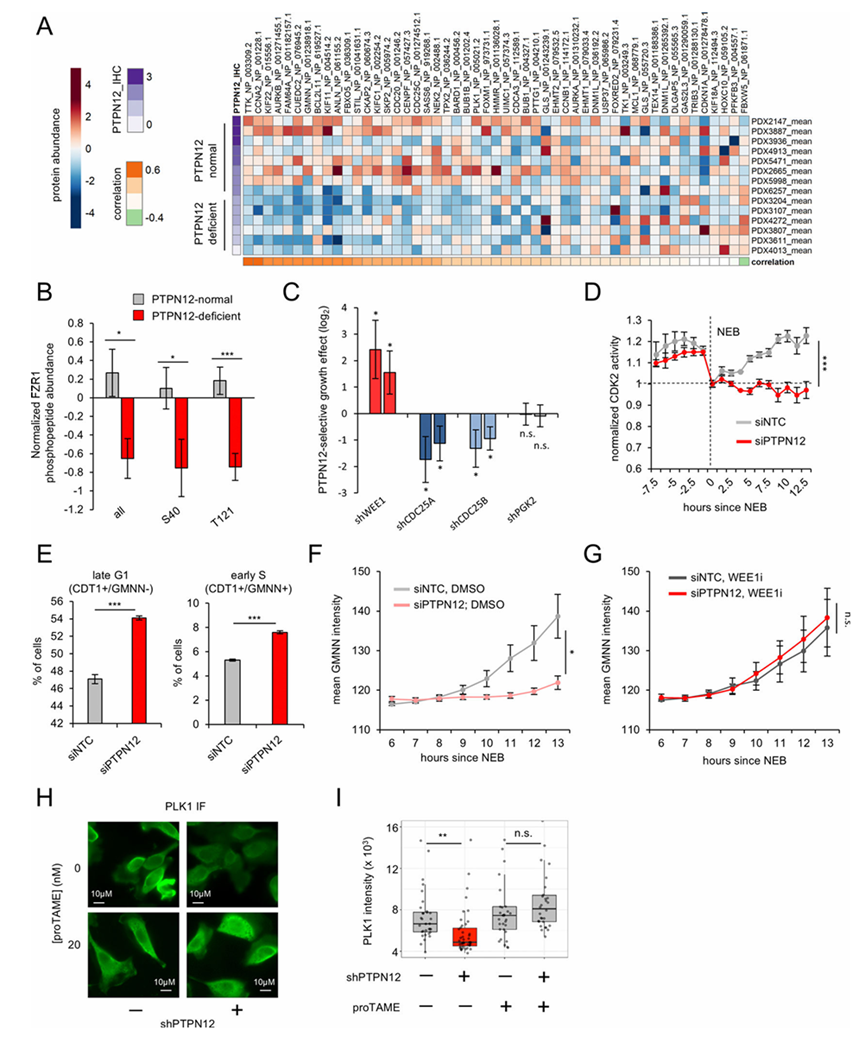
图4. PTPN12 缺乏导致 APCFZR1底物异常降解。
(A) TNBC PDX模型中,PTPN12状态与APC底物丰度相关。(B) PTPN12缺陷型PDX模型中特定FZR1残基的磷酸化水平降低。(C) CDK2活性调节因子是PTPN12选择性生长调节因子。(D) PTPN12缺失会损害CDK2的激活。(E) PTPN12失活导致G1期和早期S期细胞的积累。(F) PTPN12耗竭导致S期GMNN积累延迟。(G) PTPN12 缺陷细胞中 GMNN 的延迟积累可通过抑制 WEE1 来抑制。(H) PTPN12 缺陷导致 PLK1 异常耗竭,而 APC 抑制剂可挽救这种耗竭。(I) APC 抑制剂可挽救 PTPN12 缺陷引起的 PLK1 异常耗竭。
05
PTPN12 缺乏会导致依赖于 APC FZR1过度激活的有丝分裂异常
基于PTPN12缺陷与APC FZR1底物异常耗竭(这些底物在有丝分裂进程中起关键作用)相关的发现,他们假设PTPN12失活可能导致有丝分裂异常。为了验证这一假设,他们构建了shPTPN12-HMEC细胞,并引入H2B-GFP融合cDNA,以便通过活细胞显微镜观察凝缩的有丝分裂染色体。这些细胞经强力霉素处理24小时以诱导PTPN12敲低,随后进行24小时的成像。结果显示,PTPN12耗竭导致异常有丝分裂事件显著增加,包括中期阻滞、分离错误和纺锤体缺陷,而表达shRNA抗性PTPN12 cDNA可以挽救这些异常(图5A)。他们还检测了PTPN12是否调控TNBC细胞系的有丝分裂保真度,发现敲低PTPN12在PTPN12表达正常的MDA-MB-231细胞中会导致有丝分裂缺陷增加,而重新表达PTPN12在PTPN12缺陷的SUM159细胞中则会减少异常有丝分裂事件的数量(图5B-C)。重要的是,用纺锤体组装检查点(SAC)激酶TTK抑制剂reversine处理后,PTPN12缺陷细胞中中期阻滞频率的增加也发生了抑制(图5D)。此外,使用 proTAME 抑制 APC 或敲低 FZR1 也能挽救由 PTPN12 缺失引起的有丝分裂缺陷,这表明PTPN12 缺陷细胞中APCFZR1底物的异常缺失会导致有丝分裂保真度的下游后果(图 5E)。鉴于 PLK1 在有丝分裂进程的多个阶段发挥着关键作用,其在 PTPN12 缺陷细胞中依赖于 APC 的缺失,以及其与 PTPN12 的合成致死表型,他们假设 PLK1 的重新表达可能能够挽救由 PTPN12 失活引起的有丝分裂缺陷。事实上,他们发现,在PTPN12缺陷细胞中,通过重新表达dox诱导型PLK1 cDNA可以挽救有丝分裂缺陷,这与其他内源性PLK1耗竭的情况下观察到的有丝分裂错误挽救现象一致(图5F)。这些结果支持了PTPN12扰动导致APCFZR1异常耗竭的假设。在细胞周期的多个阶段,PTPN12 都会影响底物,包括 PLK1,而 PLK1 是 PTPN12 缺陷细胞中观察到的有丝分裂缺陷的关键因素。与这些观察结果一致,PTPN12 缺陷的 PDX 模型也显示出大量拷贝数改变,表明 PTPN12 失活引起的有丝分裂不保真性导致体内染色体不稳定 (CIN)(图5G-H)。
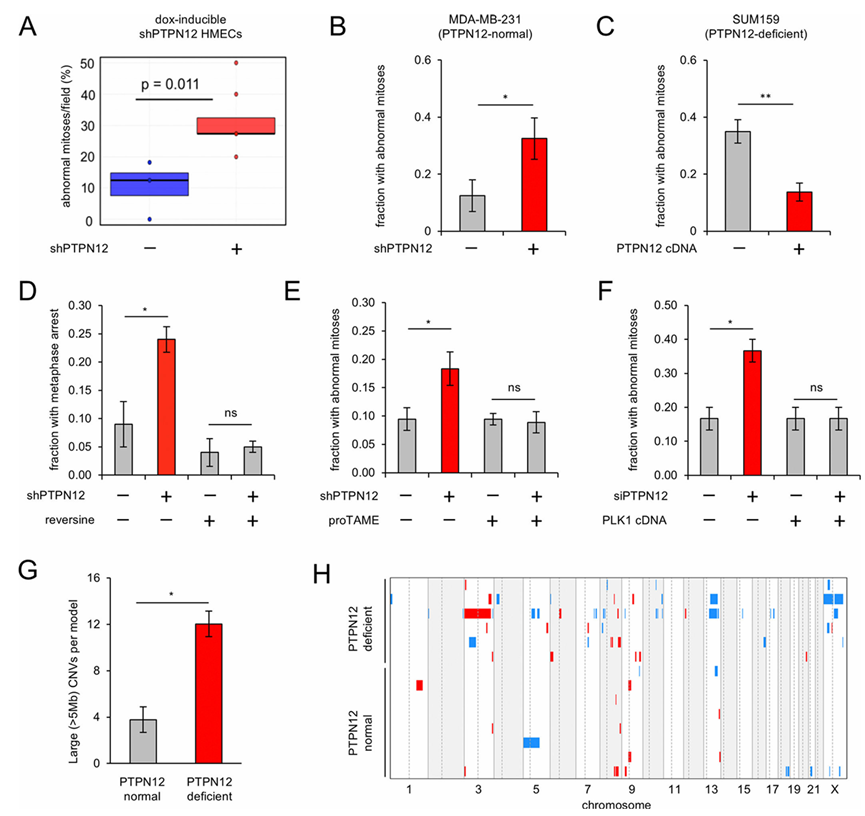
图5. PTPN12 缺陷细胞表现出依赖于 APCFZR1过度激活的有丝分裂缺陷。
(A) PTPN12 敲低导致多种有丝分裂缺陷。(B) PTPN12 敲低导致 MDA-MB-231 三阴性乳腺癌细胞有丝分裂缺陷。(C) PTPN12 的重新表达挽救了 PTPN12 缺陷型 SUM159 三阴性乳腺癌 (TNBC) 细胞的有丝分裂缺陷。(D) PTPN12 缺陷型细胞的中期阻滞是由纺锤体组装检查点 (SAC) 激活引起的。(E) APC抑制剂可挽救PTPN12缺失引起的有丝分裂缺陷。(F) PTPN12缺陷细胞的有丝分裂缺陷是由PLK1缺失驱动的。(G) PTPN12缺陷的PDX模型表现出染色体不稳定性(CIN)增加。(H) 热图展示了PTPN12表达正常和缺陷的PDX模型中大CNV的发生率和分布。
06
PTPN12 缺乏与三阴性乳腺癌患者对紫杉烷类药物的敏感性相关
本研究的数据支持这样一种模型:PTPN12缺陷细胞和肿瘤表现出APCFZR1的过度激活,导致细胞周期和有丝分裂进程缺陷,并对进一步的有丝分裂扰动更加敏感。这一观点引出了一个治疗假设:TNBC中PTPN12的缺陷可能导致对同样会引起有丝分裂应激的化疗药物(例如紫杉烷类药物)产生协同敏感性,而紫杉烷类药物是目前TNBC标准治疗方案的组成部分(图6A)。为了验证PTPN12缺陷是否与对紫杉烷类药物的敏感性相关,他们首先通过活细胞成像技术评估了shPTPN12-HMEC在多西他赛处理后的有丝分裂保真度。有趣的是,PTPN12的敲低增强了多西他赛引起的有丝分裂缺陷的频率和严重程度,而APC抑制剂即使在多西他赛处理的情况下也能抑制这些异常(图6B)。重要的是,PTPN12 缺失也降低了暴露于多西他赛的细胞的克隆形成存活率,而 PLK1 的过表达可以部分挽救这种表型,这表明观察到的有丝分裂缺陷对细胞活力具有功能性影响,并且 PLK1 缺失在 PTPN12 缺陷细胞对紫杉烷类药物的敏感性中起着关键作用。他们还测试了 PTPN12 是否能够调节 TNBC 细胞系模型中对多西他赛的敏感性,结果与之前的研究结果一致,PTPN12 敲低导致 MDA-MB-231 细胞有丝分裂缺陷增加,而 PTPN12 缺陷的 MDA-MB-231-LM2 或 SUM159 细胞中 PTPN12 的重新表达则抑制了紫杉烷类药物诱导的有丝分裂缺陷。接下来,为了评估PTPN12缺陷是否会导致TNBC临床前模型对紫杉烷类药物敏感,他们利用来自一位超早发性乳腺癌患者(诊断时23岁)的PDX模型评估了多西他赛的反应。该患者携带PTPN12的局灶性种系缺失,且缺乏其他已知的与早发性乳腺癌相关的种系改变(例如BRCA1、BRCA2、TP53、PALB2等)。值得注意的是,这些PTPN12缺陷型肿瘤在单药多西他赛治疗后表现出明显的消退,这与PTPN12失活使TNBC对紫杉烷类化疗敏感的假设相符。为了在异质性TNBC队列中验证这一假设,他们还对14个PDX模型中的11个进行了蛋白质组学分析,评估了多西他赛的反应。结果发现,PTPN12低表达的PDX模型对多西他赛的反应显著增强,大多数PTPN12缺陷型PDX模型表现出显著的肿瘤消退(图6C-D)。综上所述,这些结果支持了 PTPN12 失活是 TNBC 患者中相当一部分人对紫杉烷类药物敏感的假设,并为进一步评估 PTPN12 作为临床队列中紫杉烷类药物反应的预测生物标志物提供了强有力的理论依据。
基于这些结果,他们试图检验PTPN12状态是否可以预测TNBC患者对紫杉烷类化疗的临床反应,因为紫杉烷类药物是TNBC患者标准治疗的核心组成部分。为了评估这一点,他们使用了CALGB-9344(INT-0148)临床试验的样本。该试验旨在评估在淋巴结阳性乳腺癌女性的标准辅助化疗方案中加入序贯紫杉醇治疗的益处。该研究发现,对于ER阴性乳腺癌患者,接受紫杉醇治疗可显著降低复发风险。基于此,他们回顾性地使用经验证的IHC方法,对来自该试验约1600例患者的福尔马林固定标本中的PTPN12状态进行了评估。将这些数据与从相同样本中获得的ER和HER2评分整合后,发现27%的ER−/HER2−肿瘤也存在PTPN12缺陷(IHC≤2)(图6E)。随后,他们使用Cox比例风险模型分析比较了接受标准治疗或标准治疗联合紫杉醇治疗的PTPN12正常组和PTPN12缺陷组的临床结局。值得注意的是,他们观察到仅在PTPN12缺陷肿瘤患者中,无病生存期和总生存期均有延长的趋势,这表明PTPN12缺陷可能是TNBC中紫杉烷类药物疗效的特异性预测因子;然而,由于该分析样本量不足以检测PTPN12缺陷TNBC亚组(N=79例患者)中的效应,因此上述趋势均未达到统计学意义(图6F)。他们从细胞系、PDX 和临床样本中获得的结果共同表明,乳腺癌中 PTPN12 失活导致 CDK2 和 APCFZR1活性失调,进而导致细胞周期和有丝分裂进程的下游缺陷、有丝分裂异常率增加以及对紫杉烷类化疗等有丝分裂进一步扰动的敏感性,尽管还需要更适当的统计分析才能在临床环境中证实这一点。
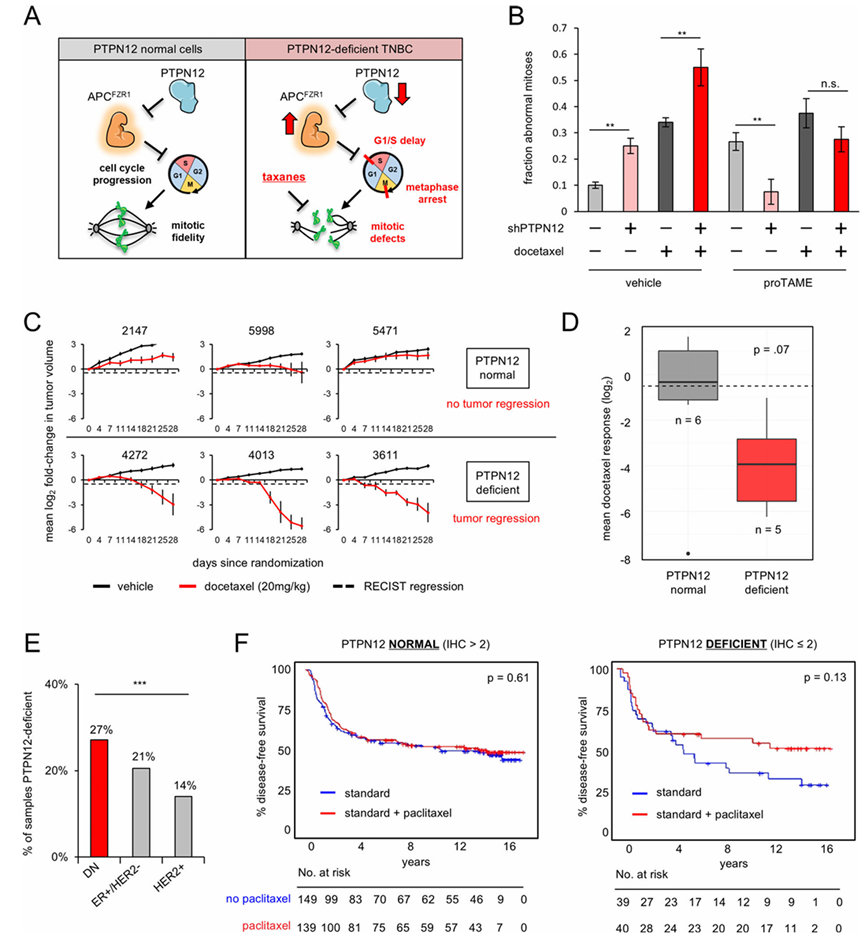
图6. PTPN12 缺陷的原发性 TNBC 对紫杉烷类化疗敏感。
(A) APCFZR1过度激活导致 PTPN12 缺陷型TNBC细胞有丝分裂缺陷。(B) APC 抑制剂可抑制紫杉烷类药物治疗引起的 PTPN12 缺陷型细胞有丝分裂缺陷的增加。(C) PTPN12 缺陷型患者来源PDX对多西他赛的反应更强。(D) PDX模型的多西他赛反应。(E) 超过25%的三阴性乳腺癌(TNBC)中PTPN12蛋白表达缺失。条形图显示了CALGB9344研究中ER+/HER2−、HER2+和双阴性(DN)亚型患者标本中PTPN12低表达(IHC≤2)的比例。(F) 在CALGB9344研究中,PTPN12缺陷型肿瘤患者从辅助紫杉醇治疗中获益更多。
+ + + + + + + + + + +
结 论
本研究通过整合蛋白质组学特征分析和合成致死性筛选,鉴定了肿瘤抑制因子 PTPN12 失活的 TNBC 中肿瘤选择性脆弱性。PTPN12 失活通过异常激活泛素连接酶复合物 APC FZR1(细胞周期的关键调控因子)驱动有丝分裂缺陷。与 PTPN12 失活在 TNBC 细胞系中引起的有丝分裂应激相一致,PTPN12 缺失的肿瘤对紫杉烷类化疗表现出更高的敏感性。总的来说,这些数据表明 PTPN12 失活可能驱动 TNBC 中的染色体不稳定性和有利的 MTA 反应。
+ + + + +



