

 English
English文献解读|Cell Rep Med(10.6):胃癌患者肠道和口腔微生物群的独特特征
✦ +
+
论文ID
原名:Distinct signatures in the human gut and oral microbiomes of gastric cancer
译名:胃癌患者肠道和口腔微生物群的独特特征
期刊:Cell Reports Medicine
影响因子:10.6
发表时间:2026.04.20
DOI号:10.1016/j.xcrm.2026.102761
背 景
胃癌(GC)是全球第五大常见癌症,无论从发病率还是癌症相关死亡率来看都是如此。东亚地区承受着不成比例的负担,占GC相关死亡人数的一半以上。幽门螺杆菌(Hp)感染是公认的GC危险因素,影响着全球超过一半的人口。然而,仅有不到5%的Hp感染者会发展为GC,且在随机对照试验中,根除Hp仅能轻微降低GC的发病率。这表明,还有其他微生物因素参与GC的发生发展,因此有必要对GC发病机制中的微生物组进行更广泛的研究。
实验设计
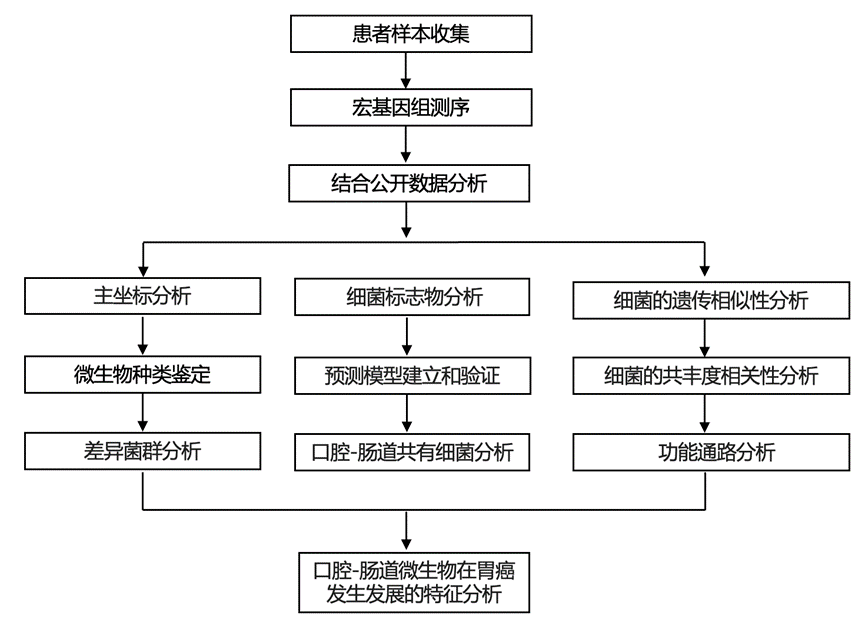
结 果
01

对 404 份粪便和唾液样本进行宏基因组测序
为了研究与GC相关的微生物标志物,研究团队对GC和慢性胃炎(ChG)患者的粪便和唾液样本进行了宏基因组测序(图1A)。粪便宏基因组数据来自两个独立的队列:队列1包含106例ChG患者和106例GC患者,队列2包含52例ChG患者和53例GC患者。两个队列的人口统计学特征,包括性别、年龄和体重指数(BMI),具有可比性。本研究产生了三个数据集:队列1的粪便宏基因组(粪便1)、队列2的粪便宏基因组(粪便2)和队列2的唾液宏基因组(唾液2)。值得注意的是,87例患者(43例ChG和44例GC)拥有配对的粪便和唾液宏基因组。
主坐标分析 (PCoA) 显示粪便和唾液微生物组之间存在明显分离,队列 1 和队列 2 的粪便样本之间没有明显的聚类(图 1B)。与粪便微生物组相比,唾液微生物组的 α 多样性更高,以香农指数和观察到的物种数衡量(图 1C)。β 多样性分析显示,GC 患者和 ChG 患者唾液微生物组的微生物组成差异大于粪便微生物组(图1D-F)。置换多元方差分析 (PERMANOVA) 表明,癌症状态比年龄、性别或 BMI更能解释粪便和唾液微生物组的变异。在校正年龄、性别和 BMI 后,GC患者和ChG患者的肠道或口腔微生物组的 α 多样性均无显著差异。
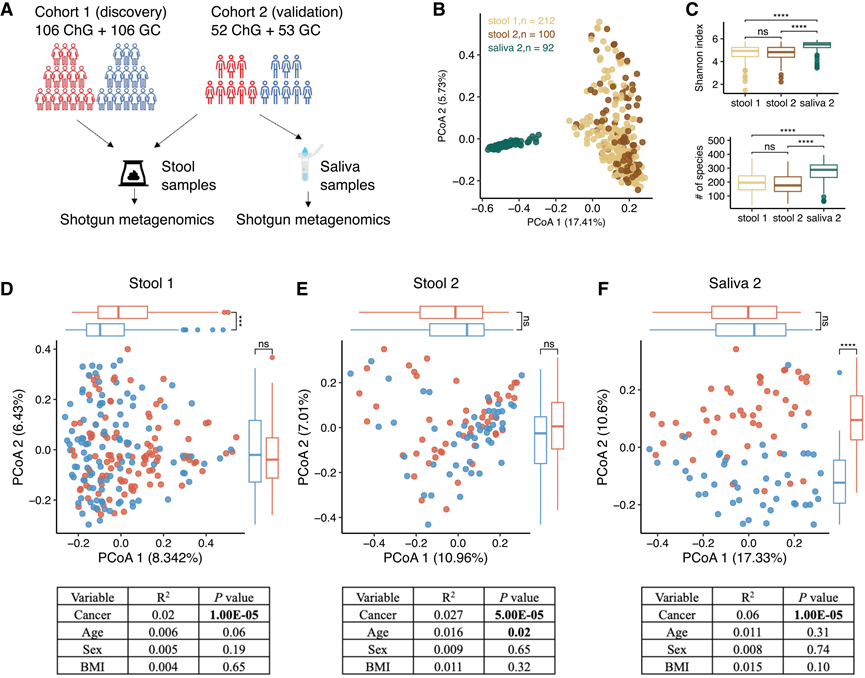
图1. 404 个粪便和唾液宏基因组的总体特征。
(A)研究设计。(B) 基于404个样本(312个粪便样本和92个唾液样本)物种丰度分布的Bray-Curtis距离的主坐标分析(PCoA)图。(C) 队列 1 和队列 2 的粪便和唾液宏基因组的 alpha 多样性(香农指数和观察到的物种)箱线图。(D) 来自队列 1 的 212 个粪便宏基因组的 PCoA。(E) 对来自队列 2 的 100 个粪便宏基因组进行 PCoA 分析。(F) 来自队列 2 的 92 个唾液宏基因组的 PCoA。
02
肠道和口腔中区分ChG和GC的微生物种类
为了鉴定与GC相关的肠道微生物物种,他们利用宏基因组数据,对队列 1 和队列 2 中的物种丰度进行了两步验证性宏基因组关联研究 (MWAS)。首先,使用三种方法(Wilcoxon 秩和检验、MaAsLin2和 ANCOM-BC)检测了队列1中 485 个物种(在 >10% 的样本中出现)的丰度与GC状态之间的关联,60个物种在GC患者和ChG患者之间存在显著差异(图 2A)。其中,28 个物种在队列2中得到验证(图 2B)。他们进一步使用MaAsLin2 分析了年龄、性别和BMI对这 28 个差异物种的潜在混杂效应。值得注意的是,调整后仍有24种物种具有显著性;其余4种不显著的物种中,有3种仅在36-73个个体中检测到,这可能是由于多变量模型的统计效力不足所致。对未经Hp治疗的样本进行的敏感性分析表明,28种物种中有27种仍具有显著差异。因此,后续分析主要集中于这28种已验证的肠道菌群标志物。
在28种差异性肠道菌群中,23种在GC患者中富集,而5种在GC患者中较ChG患者减少(图2B)。GC患者中减少的菌群包括Bacteroides ovatus和Fusicatenibacter saccharivorans等有益菌。值得注意的是,大多数GC患者富集的菌群为口腔共生菌,其中11种此前已报道为口腔-肠道传播菌。8种菌群属于链球菌属,包括咽峡炎链球菌(Sa)。Sa与GC的关联性最强。为了确定其他链球菌与GC的关联是否独立于Sa,他们进行了条件分析。五种链球菌——S. constellatus、S. gordonii、S. infantis、S. mitis和S. oralis——与GC显著相关,且这种相关性独立于Sa,凸显了链球菌在GC肠道微生物群中的重要性。此外,年龄在MaAsLin2模型中并非显著变量,表明链球菌的富集主要GC癌的标志,而非年龄的影响。其他在GC中富集的菌种包括乳酸菌(LAB),例如Ligilactobacillus salivarius、Limosilactobacillus fermentum、Bifidobacterium dentium、Rothia SGB49305、Lactobacillus gasseri、Limosilactobacillus oris和Limosilactobacillus vaginalis。基于基因组相似性,Limosilactobacillus和Ligilactobacillus是最近从乳杆菌属重新分类的属。由于链球菌属属于LAB,这些发现与先前关于GC胃微生物群中乳酸菌富集的报道相符。
在唾液微生物组中,他们鉴定出36种细菌在GC和ChG患者之间存在差异(图2C)。最显著的关联是Prevotella nanceiensis在GC患者中的丰度降低。Sa在GC患者中的丰度略高于ChG患者。与肠道微生物组不同,唾液中大多数差异菌种在GC患者中减少,仅有7种菌种增多(图2D)。值得注意的是,唾液中36种差异菌种中有22种,粪便中28种差异菌种中有10种在至少50%的患者中存在,这表明GC相关的唾液微生物组变化涉及更常见的菌种,而肠道微生物组变化涉及更罕见的菌种。他们在哈尔滨内部队列中验证了这些发现。虽然唾液和舌部微生物组存在固有差异,但36个差异菌种中有18个经统计学验证,其中30个显示出方向一致的变化。
接下来,他们评估了这些细菌标志物对GC状态的预测效用。在28种鉴别粪便菌种中,Boruta鉴定出17种比随机探针更具相关性(图3A)。这17种菌种包括10种口-肠传播菌,其中7种的重要性排名最高,凸显了它们与疾病的相关性。基于这17种标志物的分类器,采用六种方法进行评估,在测试数据中获得了0.76-0.85的受试者工作特征曲线下面积(AUROC),其中随机森林模型表现最佳。基于队列1粪便样本训练的随机森林模型预测队列2粪便样本的AUROC为0.85(图3B),准确率为0.80(图3C)。在36种差异性唾液菌种中,有20种的相关性高于随机探针(图3D)。随机森林模型在训练和测试中均表现出色。基于第二组唾液样本训练的模型对哈尔滨舌样本的预测AUROC为0.87(图3E),准确率为0.82(图3F)。该唾液模型包含Corynebacterium matruchotii,该菌是最具预测性的菌种。Wilcoxon检验和ANCOM-BC检验均支持Corynebacterium matruchotii丰度与GC之间的关联,但MaAsLin2检验未发现这种关联。
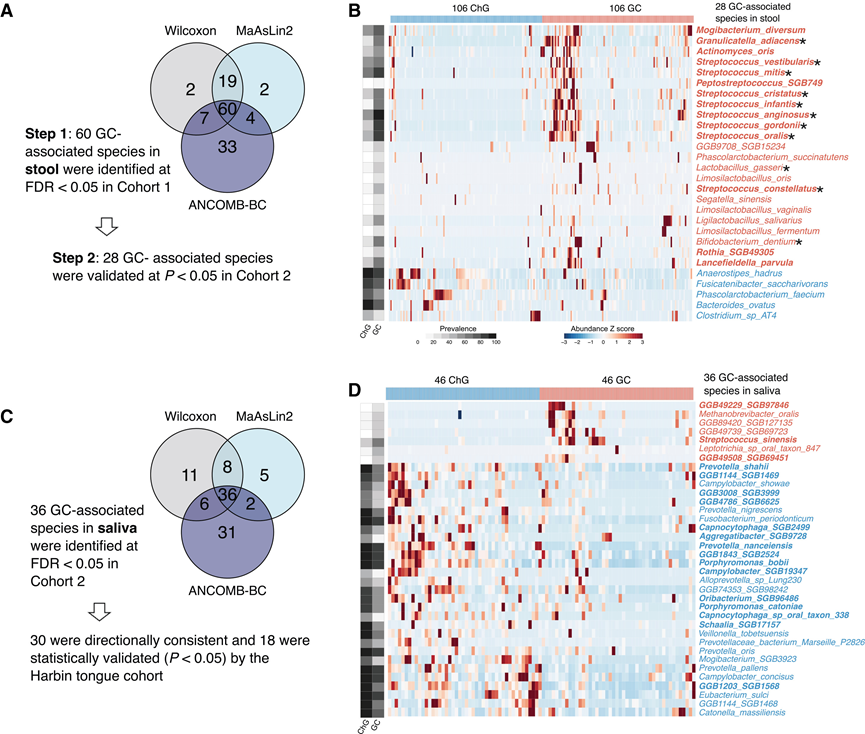
图2. 肠道和口腔中GC组和ChG组之间丰度不同的微生物种类。
(A) 粪便样本中物种水平差异分析的两步验证框架。(B) 来自队列 1 的 212 个粪便宏基因组中,GC 和 ChG 之间 28 种差异丰度物种的热图。(C) 口腔样本中物种水平差异分析的统计框架。(D) 来自队列2的92个唾液宏基因组中,GC组和ChG组之间36种差异表达物种的热图。
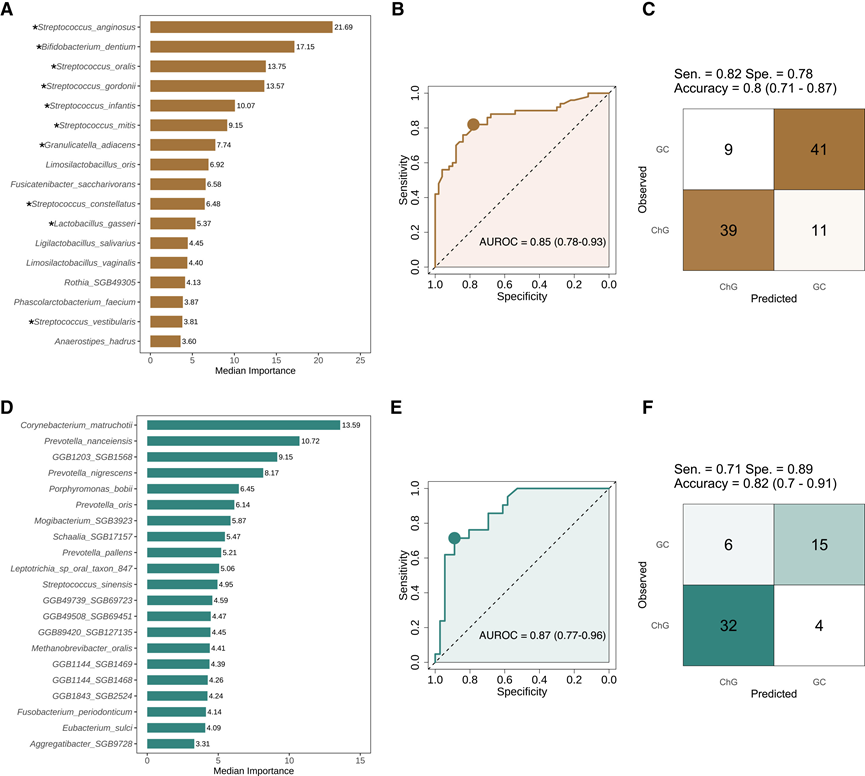
图3. 肠道和口腔细菌标志物对GC的预测性能。
(A) 根据 Boruta 算法鉴定的粪便微生物物种标志物,按重要性评分中位数排序。(B) 随机森林分类器的受试者工作特征曲线(ROC曲线)。(C) (B)中最佳分类的混淆矩阵。(D) 通过 Boruta 算法鉴定的唾液微生物物种标记物,按中值重要性得分排序。(E) 随机森林分类器的ROC曲线。(F) (E)中最佳分类的混淆矩阵。
03
肠道中富含GC的物种主要为口腔-肠道共有物种
为了研究GC患者的口腔-肠道微生物特征,他们利用队列2中匹配的口腔和肠道宏基因组分析了共有微生物物种。为了尽量减少稀有分类群的影响,过滤了在任何数据集中出现频率≤5%的物种,最终保留了1163个物种。其中,206个物种存在于所有三个数据集(粪便1、粪便2、唾液2)中(图4A)。这些共有物种中约有一半属于厚壁菌门(Firmicutes phylum),其次是放线菌门和糖杆菌门(图4B)。在属水平上,链球菌属(Streptococcus)的代表性最高(19个物种),其次是放线菌属(Actinomyces)(图4B )。他们将这206个物种归类为口腔-肠道共有菌,其余物种归类为非共有菌。结果显示,匹配的唾液-粪便样本对之间的菌株相似性明显高于个体间唾液宏基因组之间的菌株相似性。
共有菌种在口腔和肠道微生物群中表现出不同的流行率和丰度模式。在唾液中,共有菌种的中位流行率为 59.8%,显著高于非共有菌种(图 4C)。相比之下,在肠道中,大多数共有菌种在不到12%的患者中出现,流行率低于非共有菌种。共有菌种对唾液中微生物总丰度的贡献率为 59.6%,但在粪便中仅为 6.4%(图 4D)。值得注意的是,在肠道中,与 ChG 患者相比,GC 患者的共有菌种流行率显著更高,而在唾液中未观察到这种差异。
接下来,他们将差异菌种分为共有菌种和非共有菌种两类。在唾液微生物组中,36种差异菌种中仅有7种为共有菌种,大多数GC含量降低的菌种属于非共有菌种组(图4E)。在肠道微生物组中,28种差异菌种中有20种为口腔-肠道共有菌种,且这些菌种均富含GC(图4E)。这20种富含GC的共有菌种在肠道中的丰度显著高于其他共有菌种(图4F)。这些发现凸显了口腔-肠道共有菌种在驱动GC相关肠道微生物组改变中的重要作用。
为了检验富含GC的肠道菌群是否来源于口腔,他们利用匹配的唾液和粪便宏基因组进行了菌株水平分析。鉴于肠道中共有菌群的流行率和丰度较低(图5A),他们重点关注了8种在粪便中流行率>30%的链球菌,这些菌群此前已鉴定为口-肠传播菌,且在唾液中的丰度高于粪便:S. anginosus、S. constellatus、S. cristatus、S. gordonii、S. infantis、S. mitis、S. oralis和S. vestibularis。群体平均核苷酸同一性(popANI)(一种衡量菌株相似性的指标)在同一受试者的匹配唾液和粪便宏基因组之间的差异显著高于不同受试者的唾液样本之间的差异(图5B)。具体而言,42%-83%的匹配比较结果显示popANI>0.999,而个体间唾液比较结果中仅有<3%达到此标准。在87名具有匹配宏基因组的个体中,44名(23名GC患者,21名ChG患者)至少对一种链球菌属具有可检测的popANI,且与其余个体相比,年龄差异不显著。年龄与popANI之间的物种相关性较弱(图5C),表明口-肠传播不受年龄的影响。这些结果支持GC患者肠道中这些链球菌属的口源性,并证实了它们作为口-肠传播媒介的作用。
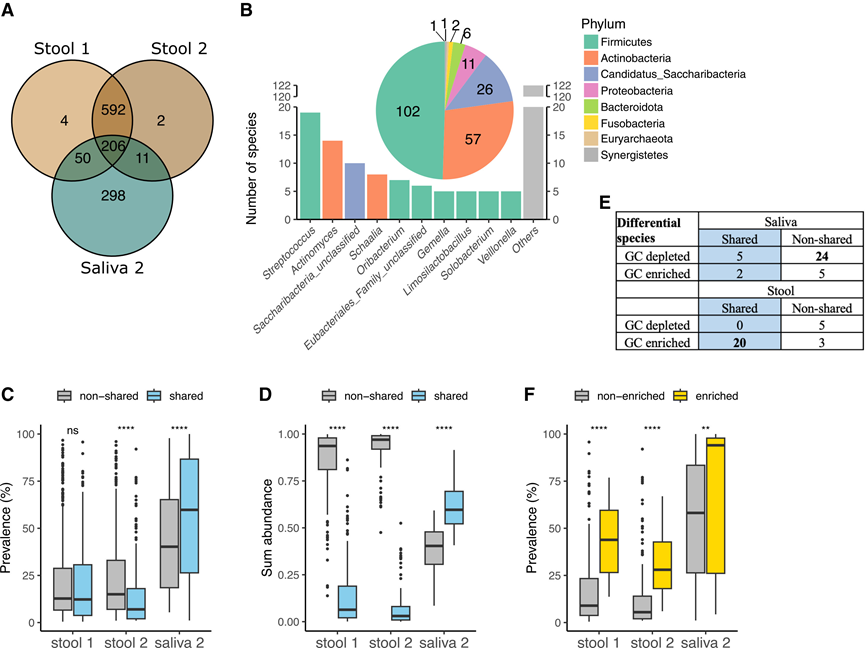
图4. 富含 GC 的肠道微生物群中口腔-肠道共有物种的优势地位。
(A) 维恩图显示了在粪便(队列 1 和队列 2)和唾液(队列 2)宏基因组中检测到的物种数量。(B) 在所有数据集中检测到的206个口腔-肠道共有物种的门和属组成。(C) 粪便和唾液宏基因组中口腔-肠道共有物种与非共有物种的患病率箱线图。(D) 粪便和唾液宏基因组中口腔-肠道共有物种与非共有物种丰度总和的箱线图。(E) 按口肠道共有状态分层的差异丰度物种。(F) 粪便中富含GC与未富含GC的口腔-肠道共有菌种的患病率箱线图。
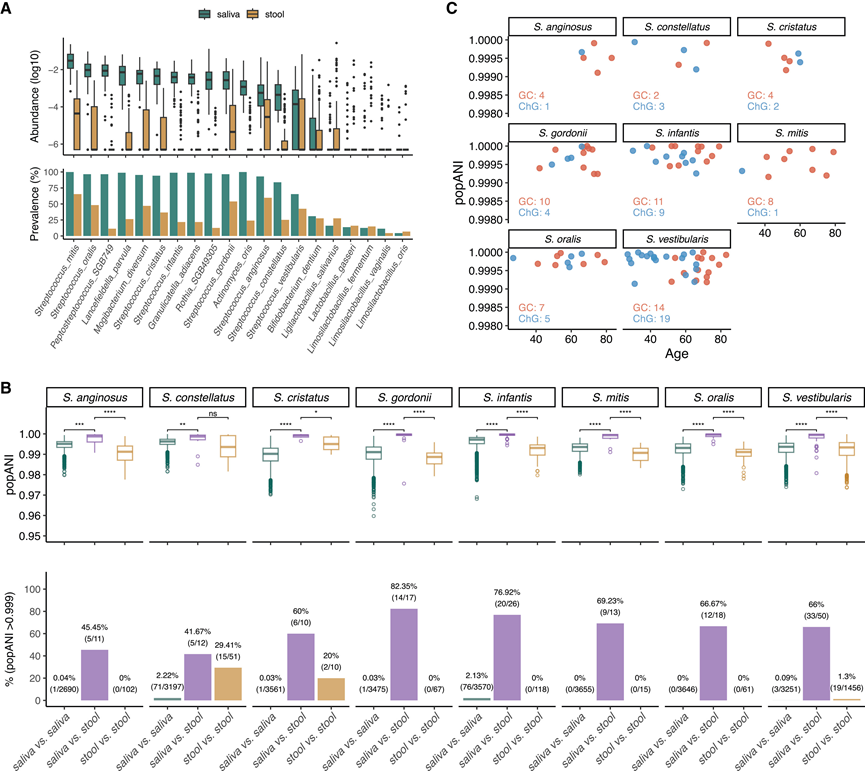
图5. 粪便和唾液微生物组中富含 GC 的口腔-肠道共有物种的遗传相似性。
(A) 队列 2 中 20 种富含 GC 的口腔-肠道共有菌种的丰度和流行率。(B)8种富含GC的口腔-肠道共有链球菌的群体平均核苷酸同一性(popANI)箱线图,比较了(1)来自同一受试者的匹配粪便和唾液宏基因组,(2)来自不同受试者的唾液宏基因组,以及(3)来自不同受试者的粪便宏基因组。(C) 根据匹配的唾液和粪便宏基因组分析出的8种链球菌的年龄与 popANI 的散点图。
04
肠道中富含GC的物种在口腔和肠道微生物组中均表现出很强的共丰度相关性
同一生态位内的微生物群落通常表现出密切的相互作用,共丰度网络可以揭示生物学见解。为了探索GC中的微生物相互作用,他们使用FastSpar构建了口腔和肠道微生物组的共丰度网络。在粪便和唾液微生物组中,口腔-肠道共有菌种的共丰度相关性比非共有菌种更频繁且更强(图6A)。对于共有菌种,在粪便1、粪便2和唾液2数据集中,分别有20.9%、7.3%和8.7%的可能相互作用具有显著性,而非共有菌种的相应比例分别为8.0%、1.8%和3.5%。在共有菌种中,GC富集菌种的相关性显著高于GC含量低或无差异的共有菌种(图6B)。具体而言,在粪便1、粪便2和唾液2数据集中,GC富集菌种的相应比例分别为74.7%、36.8%和18.9%,而其他共有菌种的相应比例分别为20.5%、7.1%和8.6%。
他们着重分析了队列1和队列2所有三个数据集中富含GC的物种之间的稳健共丰度网络,并鉴定出15个涉及14个物种的正相关性(图6C)。他们观察到乳酸菌(LAB)之间存在紧密联系的网络,包括Limosilactobacillus oris、Limosilactobacillus vaginalis、Limosilactobacillus fermentum、Lactobacillus gasseri和Bifidobacterium dentium,该网络延伸至S. gordonii、Ligilactobacillus salivarius和Rothia SGB49305。此外,Lancefieldella parvula与Actinomyces oris、Mogibacterium diversum和Schaalia meyeri相关。值得注意的是,在哈尔滨舌标本数据中,15个正相关性中有10个得到了验证(图6C)。
鉴于LAB在GC中的富集,他们分析了粪便宏基因组中的乳酸发酵通路。同型乳酸发酵通路(ANAEROFRUCAT-PWY)和异型乳酸发酵通路(P122-PWY)在队列1的GC患者中显著富集,且在队列2中方向一致。队列2中未观察到显著性可能是由于样本量较小。在对未接受Hp治疗的样本子集进行敏感性分析时,P122-PWY通路的富集仍然显著。值得注意的是,参与异型发酵通路的七种酶在队列1的GC患者中均显著富集(图7A-C)。这些酶包括影响乳酸生成的关键酶:磷酸酮酶(4.1.2.9)和L-乳酸脱氢酶及D-乳酸脱氢酶(1.1.1.27和1.1.1.28)。在同型发酵通路中,检测到的九种酶中有七种在队列1的GC患者中显著富集。这些发现与LAB的富集相一致,进一步证实了它们在GC相关肠道菌群失调中的作用。
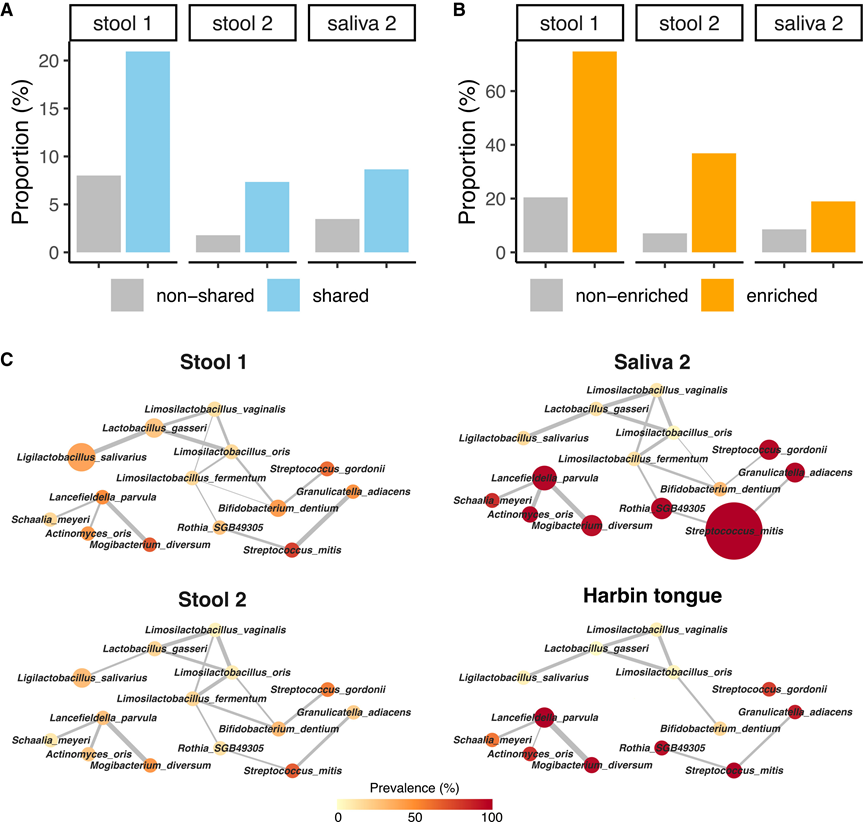
图6. 富含 GC 的口腔-肠道共有物种之间存在显著的共丰度相关性。
(A) 柱状图显示了粪便 1、粪便 2 和唾液 2 数据集中口腔-肠道共有物种与非共有物种的显著共丰度相关性比例。(B) 柱状图显示了粪便 1、粪便 2 和唾液 2 数据集中富含 GC 的口腔-肠道共有物种与未富含 GC 的口腔-肠道共有物种之间显著共丰度相关性的比例。(C) 使用 Cytoscape 可视化了四个数据集中 14 种富含 GC 的口腔-肠道共有菌种的共丰度网络。
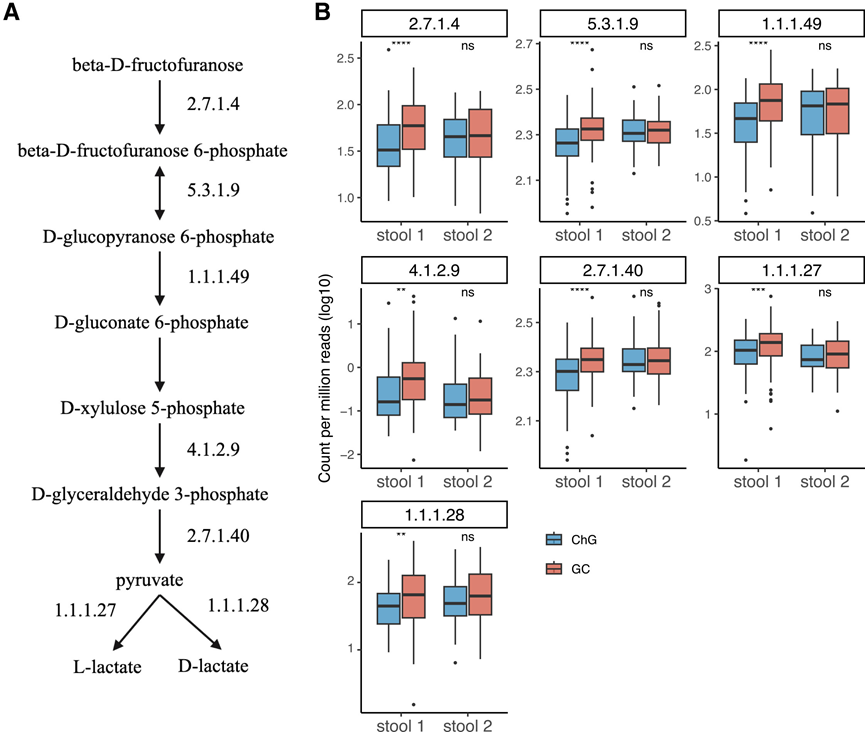
图7. GC患者粪便微生物组中异型乳酸发酵酶的富集。
(A) MetaCyc途径P122-PWY的示意图:异型乳酸发酵。(B) 箱线图显示了 GC 和 ChG 患者粪便微生物组中 7 种酶的丰度。
+ + + + + + + + + + +
结 论
本研究分析了来自两个独立队列受试者的肠道和口腔宏基因组数据,并在哈尔滨队列中进行了验证,鉴定出20种在GC患者肠道中富集的口腔-肠道共有菌种,主要为LAB。虽然大多数肠道微生物标志物在唾液中含量丰富,但在GC患者中均未发生显著改变。对87个匹配的唾液-粪便宏基因组进行菌株水平分析,证实了链球菌属的口腔-肠道传播。在GC患者中富集的乳酸菌在口腔和肠道微生物群中形成了稳定的共丰度网络,提示存在协同作用。功能分析显示,GC患者粪便中乳酸发酵通路富集,这与LAB的优势地位以及先前关于胃肠道菌群的研究结果相一致。此外,基于微生物组的分类器在GC诊断中实现了较高的预测准确率,凸显了其转化应用潜力。总而言之,这些发现强调了口腔-肠道微生物轴在GC发生发展中的关键作用。
+ + + + +



