

 English
English文献解读|Cancer Discov(33.3):多灶性肝细胞癌的肝内微生物异质性及其与宿主基因组和转录组改变的关系
✦ +
+
论文ID
原名:Intrahepatic Microbial Heterogeneity in Multifocal Hepatocellular Carcinoma and Its Association with Host Genomic and Transcriptomic Alterations
译名:多灶性肝细胞癌的肝内微生物异质性及其与宿主基因组和转录组改变的关系
期刊:Cancer Discovery
影响因子:33.3
发表时间:2025.08.04
DOI号:10.1158/2159-8290.CD-24-1259.
背 景
肝癌是全球癌症相关死亡的第三大原因,其中肝细胞癌 (HCC) 是最常见的肝癌类型,约占原发病例的 80%。约 41% 至 75% 的 HCC 患者在初次诊断时表现为多灶性肿瘤。多灶性 HCC 可能来自原发性 HCC 的肝内转移 (IM) 或来自独立克隆的多中心发生 (MO)。虽然手术切除是 HCC 的根治性治疗选择,但 HCC 的多灶性增加了手术难度和术后复发的风险。微转移是复发和远处转移的重要诱因,尤其是在 IM-HCC中,这表明与 MO-HCC 相比预后较差。然而,多灶性肝细胞癌的分子特征和微环境因素仍未得到充分研究,这阻碍了治疗决策和患者预后。
实验设计
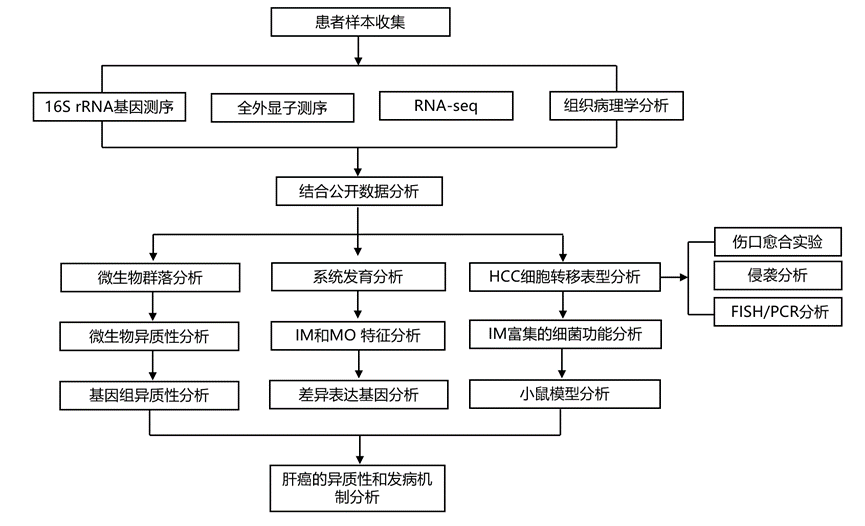
结 果
01

多灶性肝细胞癌的微生物群落特征
研究团队共收集了58例多灶性肝细胞癌(HCC)患者的300份肝组织样本,包括242个肿瘤结节样本(每例患者3-11个结节)和58个的癌旁非肿瘤肝组织样本。样本同时进行16S rRNA基因测序、全外显子测序(WES)和转录组分析(RNA-seq),以阐明多灶性HCC的微生物组、基因组和转录组特征,并进行整合分析和功能验证(图1A)。
微生物群落的整体表型模式表明,肿瘤组织和相应的邻近非肿瘤组织之间的微生物多样性(α 和 β 多样性)没有显著差异。该结果与先前的研究一致,该研究表明肿瘤旁组织的微生物负荷与肿瘤相似。接下来,他们分析了所有58例患者每个肿瘤结节及其邻近正常样本的细菌组成,以概述个体微生物群落的概况。在门水平(图1B),多灶性肝癌患者的微生物群以变形菌门(平均38.38%)为主,其次是厚壁菌门(平均17.55%)和放线菌门(平均11.45%)。在属水平,最丰富的前三个属分别是不动杆菌属(平均2.87%)、链球菌属(平均2.56%)和乳酸杆菌属(平均2.14%)。聚类分析显示,只有 3% 的患者(58 名患者中的 2 名)在肿瘤结节中表现出相似的微生物组成,而大多数患者在其肿瘤间微生物组中表现出显著的异质性,这表明多灶性 HCC 患者肿瘤结节内的微生物存在个体内差异。
为了证实多灶性 HCC 组织中存在细菌,他们对肿瘤结节进行了革兰氏染色和脂多糖(LPS,革兰氏阴性菌)和脂磷壁酸(LTA,革兰氏阳性菌)的 IHC 染色(图 1C)。肿瘤内细菌定位于核周区域,呈点状,每平方毫米约含 108 个细菌,约占肿瘤组织面积的 10.1% ± 7.5% 和 6.7% ± 4.9% (图 1C)。高分辨率电子显微镜观察发现,细菌主要分布在HCC样本的细胞质中(图1D),证实了多灶性HCC组织中存在细菌。
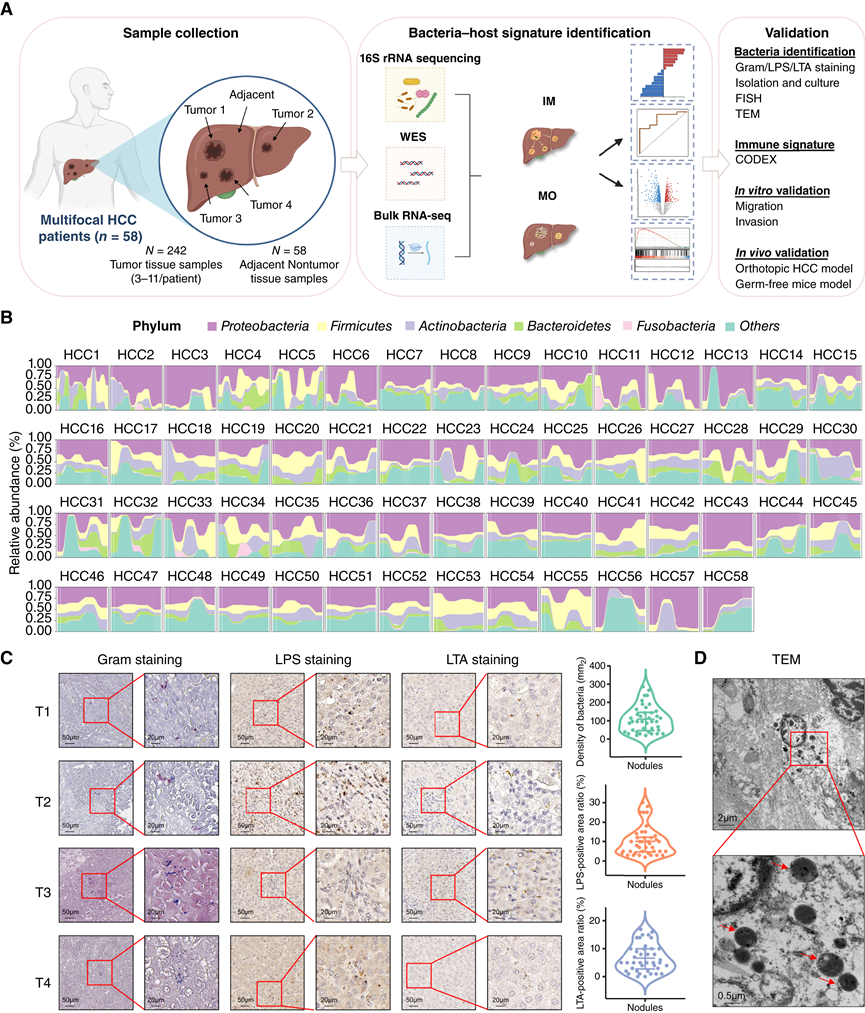
图1. 多灶性肝细胞癌的微生物分布。
(A) 样本采集和研究设计流程图。(B) 冲积图描绘了每位患者门级别(前 5 名)微生物组成的相对丰度。(C) 多灶性肝细胞癌肝组织中革兰氏染色、细菌 LPS 和 LTA 染色的代表性图像。(D) 透射电子显微镜 (TEM) 的代表性图像,显示多灶性肝细胞癌 (HCC) 肝组织内的细胞内细菌。
02
多灶性肝细胞癌中的异质性微生物群落
鉴于个体内肝细胞癌结节间微生物组成的高度变异,他们为每位患者确定了一组高变异微生物 (HVM)。根据以下三个标准选择代表性属:肝细胞癌结节与相应邻近非肿瘤组织之间属水平的微生物标准化丰度≥1,肝细胞癌结节与邻近非肿瘤组织之间的丰度差异≥0.1%,以及肝细胞癌结节间的变异系数百分比≥50。鉴定了 347 个结节间 HVM;每位患者平均有 94 个 HVM。其中,链球菌(患病率为 91.4%)、寡养单胞菌(86.2%) 和乳酸杆菌(82.8%) 是多灶性肝细胞癌患者中最普遍的 HVM。符合 HVM 标准的菌属用于为每个 HCC 组织构建微生物图谱。值得注意的是,观察到同一患者的不同 HCC 结节中 HVM 的丰度存在显著差异,网络图谱分析显示每个结节也具有其独特的 HVM(图 2)。这些发现共同推断,同一多灶性 HCC 患者的肿瘤结节中存在异质性微生物群落。
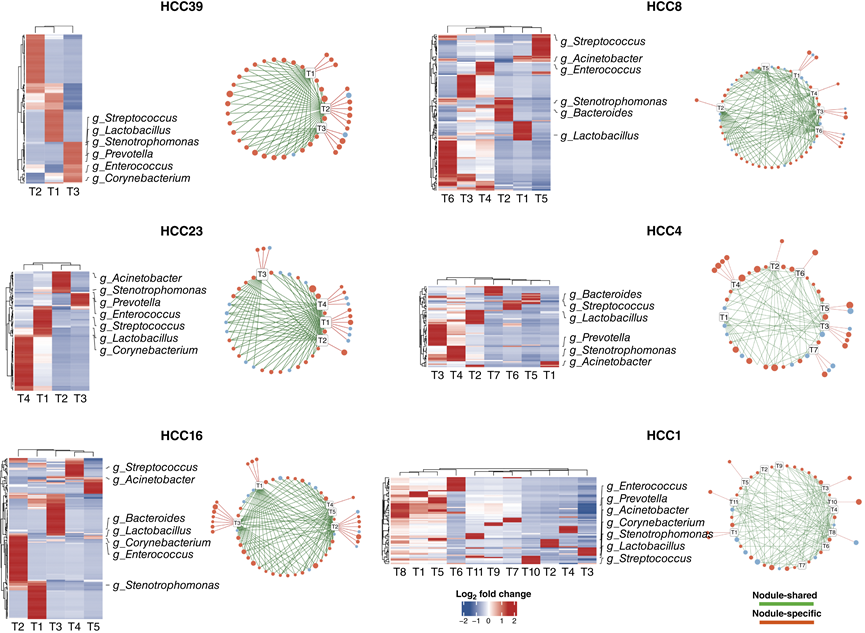
图2. 多灶性肝细胞癌中的异质性微生物群落。
03
多灶性肝细胞癌的基因组异质性与微生物异质性相关
为了探索肿瘤演进过程中可能出现的宿主基因突变,他们对56例多灶性肝细胞癌患者的235个肿瘤结节和56个癌旁非肿瘤组织样本进行了全外显子测序(WES),鉴定出29226个非同义突变和11014个同义突变,并通过桑格测序验证,真实发现率为95.3%。关注HCC驱动突变,TP53(34%)、PCLO(16%)和CTNNB1(16%)是本研究队列中最常见的突变基因(图3A)。接下来,他们通过计算Jaccard指数[肿瘤间异质性(ITH)指数]来测量个体内肿瘤间基因组异质性。结果显示,超过半数患者表现出较高的ITH水平(图3A)。这些结果提示,多灶性HCC的肿瘤间基因组突变呈现出明显的异质性。
为了判断多灶性 HCC 是起源于原发性 (IM) 还是多中心起源 (MO),他们首先将每个患者的非同义体细胞突变分为主干突变(存在于所有结节中)、分支突变(部分结节共有)或私有突变(仅存在于一个结节中)(图 3B)。同时,他们构建了系统发育树并计算了每个患者的遗传相似性(欧几里得距离),结果显示多灶性 HCC 患者可分为 MO-HCC(n = 5,无共有突变,遗传相似性极低)、IM-HCC [n=40,主干突变发生率高(8%–90%),遗传相似性强] 和混合 IM/MO-HCC(n = 11,分支突变和特有突变的组合)(图 3B)。使用 PyClone 算法进一步分析了多灶性 HCC 的克隆动态,证实了 IM-HCC 中存在常见的克隆聚类,而 MO-HCC 中不存在。他们还比较了 IM-HCC (IM) 中的 IM 结节、混合 IM/MO-HCC (MIX_IM) 中的部分 IM 结节、混合 IM/MO-HCC (MIX_MO) 中的部分 MO 结节以及 MO-HCC (MO) 中的 MO 结节的肿瘤突变负担 (TMB)。MO 结节的 TMB 高于其他结节类型,尤其是 MIX_MO和 MIX_IM。IM-HCC 患者与 MO-HCC 患者的临床特征(包括年龄、性别、体质指数、HBV 状态、HCV 状态、丙氨酸氨基转移酶、天冬氨酸氨基转移酶、总胆红素 (TBil) 和舒张胆红素 (DBil))均无显著差异。这些发现提示,同一多灶性 HCC 患者的多个肿瘤可能表现出不同的进化模式。仅对原发灶(或任何单个灶)进行测序无法完全表征多灶性 HCC 的基因组图谱。
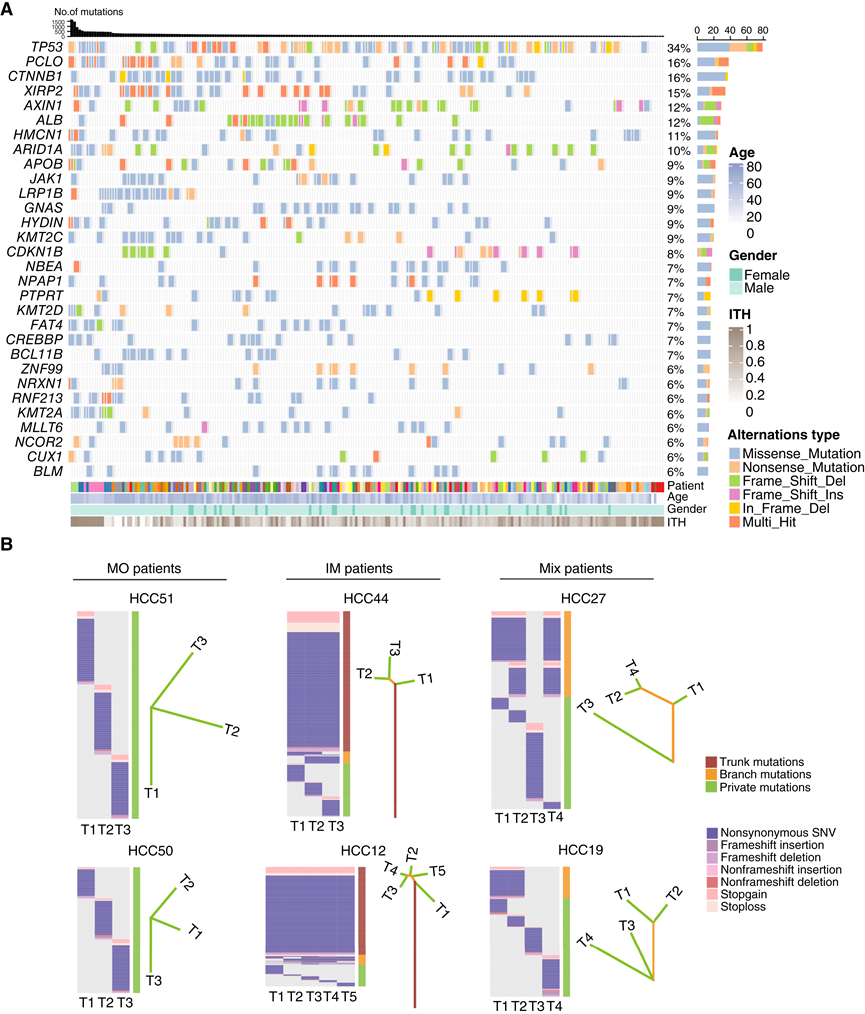
图3. 多灶性肝细胞癌的肿瘤间遗传异质性。
(A) 56 例多灶性肝细胞癌患者所有肿瘤区域的前 30 个潜在驱动基因的突变情况。(B) 6 例代表性多灶性肝细胞癌患者的突变和进化谱。
04
IM-HCC 和 MO-HCC 中不同的肿瘤内微生物群
在证明多灶性 HCC 可能具有不同的进化路径之后,他们试图研究不同类型的结节之间肿瘤内微生物群落组成是否不同。这四种类型的结节之间的微生物群落(β 多样性)存在显著差异(图 4A),尤其是在 IM 和 MO 结节之间。此外,与 MO 结节相比,IM 结节中的 α 多样性(Shannon 和 Chao1 指数)较低(图 4B)。IM/MO 结节与其相应的邻近非肿瘤组织之间的微生物群落没有显著差异(图 4C-D)。与 MO 结节相比,肠球菌属和梭杆菌属在IM结节中富集,而寡养单胞菌属、棒状杆菌属、伯克霍尔德菌属和梭菌属则发生耗尽(图 4E)。在种水平上,与 MO结节相比,小链球菌和咽峡炎链球菌在IM结节中也富集(图 4E)。为了评估细菌标记是否可以区分IM结节和 MO 结节,他们以 6:4 的比例将样本随机分为训练组和验证组。使用九个差异丰富的分类单元,构建了一个重复五倍交叉验证的随机森林模型。该模型在区分IM结节和MO结节时,AUC 达到 0.795(图 4F)。由于病例数量不平衡,他们还进行了精确召回率 (PR) 曲线下面积分析,以尽量降低假阳性率。PR 分析证实,这九种标记物可以有效地区分IM和MO,AUC 为 0.831。总之,这些结果表明,在多灶性肝细胞癌患者的IM结节和MO结节中存在不同的微生物群。
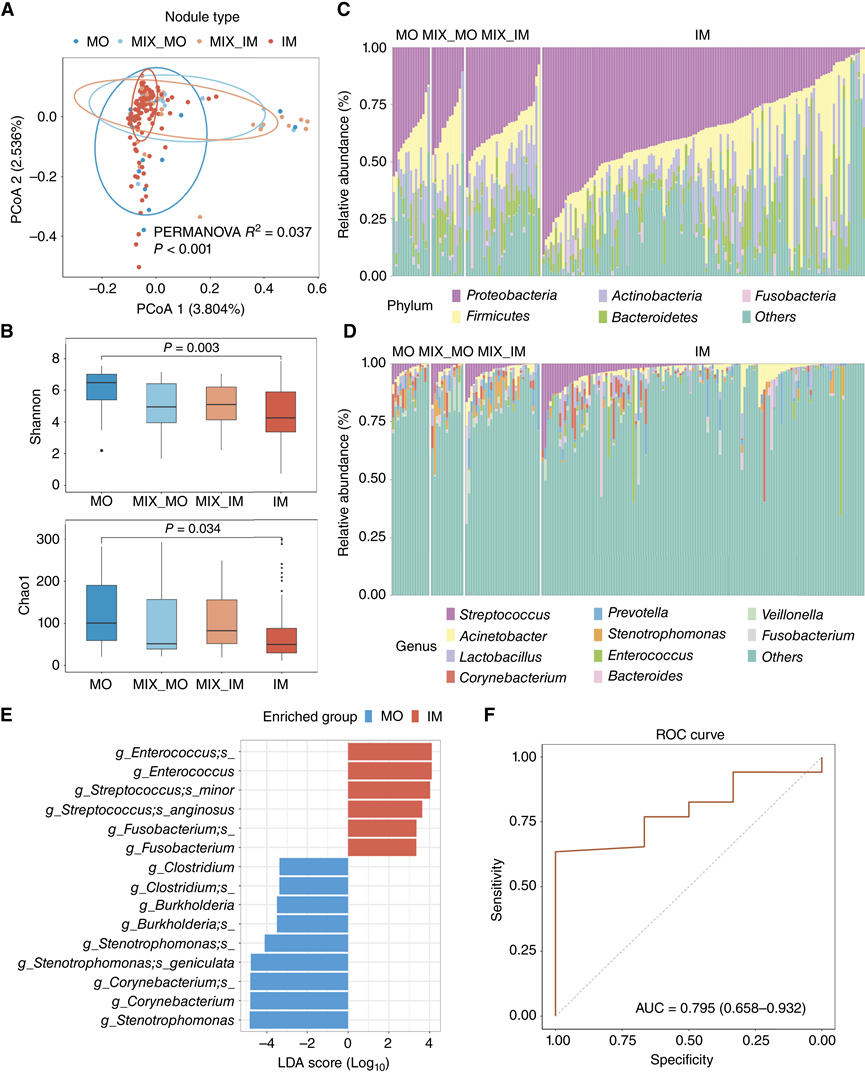
图4. 多灶性肝细胞癌IM和MO结节中微生物组的特征。
(A) 基于Bray-Curtis距离的主坐标分析(PCoA),显示MO、MIX_MO、MIX_IM和IM结节组织中的微生物群落结构。(B) 基于Shannon和Chao1指数的α多样性箱线图,比较了MO、MIX_MO、MIX_IM和IM结节组织。(C-D) 堆叠柱状图描绘了MO、MIX_MO、MIX_IM和IM结节组织中门和属组成的相对丰度。(E) 线性判别分析有效大小(LEfSe)分析显示IM和MO结节之间在属和种水平上差异丰富的分类单元。(F) ROC 曲线评估了使用重复五折交叉验证的随机森林模型,评估了九个差异丰度分类单元在区分 IM 和 MO 方面的预测能力。
05
IM肿瘤结节显示增强的转移潜能
他们进一步对收集的样本进行了 RNA-seq,以阐明IM结节和MO结节之间功能基因表达的改变,总共鉴定出 615 个差异表达基因,其中与MO结节相比,IM结节中有 434 个基因上调,181 个基因下调(图 5A)。值得注意的是,许多转移相关基因,包括AGR2、HOXB9和CEACAM6,在IM结节中显著上调。基因集富集分析显示,上皮间质转化 (EMT) 通路成为IM结节中最富集的通路,其次是细胞周期相关通路,如 G2-M 和 E2F 信号,以及血管生成(图 5B),表明IM结节具有更高的转移能力。
为了探究肿瘤内微生物是否与宿主基因表达相关,他们利用 Spearman 相关性来表征细菌与宿主基因之间的关联。他们鉴定出 3622 个基因和 139 个分类单元在IM结节中表现出显著关联。有趣的是,与IM富集分类单元(例如肠球菌属、咽峡炎链球菌、梭杆菌属和小沙门氏菌)显著相关的宿主基因与 EMT 相关(图 5C),这与 MO 结节中的关联模式不同。具体而言,小沙门氏菌与促进转移的转录因子 (SNAI2) 呈正相关,而与紧密连接标志物闭合蛋白 (OCLN)呈负相关。咽峡炎链球菌与SPARC/骨粘连蛋白、cwcv、kazal样结构域蛋白聚糖1 (SPOCK1) 和细胞粘附分子 (CEACAM) 呈正相关。另一方面,肠球菌属与葡萄糖转运蛋白 (SLC2A1) 和VEGFA(两者均为EMT中已确定的驱动因子)(图5D)相关。综上所述,这些分析表明,IM富集的致病菌可能通过激活HCC中的EMT相关信号通路参与IM。
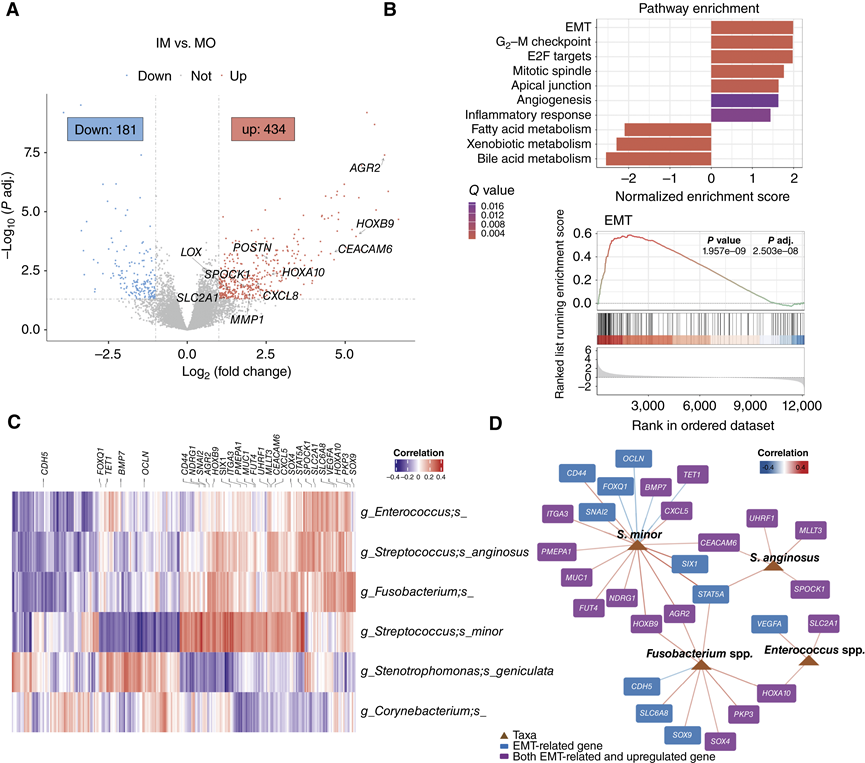
图5. IM 肿瘤表现出增强的转移活性,并且与肿瘤内微生物群相关。
(A) 火山图显示IM 和 MO 肿瘤结节之间的差异表达基因。(B) 基因集富集分析表明,在 IM 和 MO 结节之间差异调控的标志性信号通路。(C) 热图显示了通过 Spearman 相关性在 IM 结节中识别的差异微生物类群(行)和宿主基因(列)之间的相关性模式。(D) 网络分析显示 IM 结节中富集的肿瘤内微生物及其与转移相关的宿主基因的相关性。
06
IM富集的细菌促进HCC细胞转移表型
为了探究肠球菌富集菌在促进肿瘤转移中的潜在功能,他们选择了肠球菌和咽峡炎链球菌等肠球菌富集菌种进行功能验证。经过种级鉴定,将粪肠球菌选为肠球菌属的代表菌,显示其在IM结节中存在率较高。首先,他们使用特异性探针针对这些致病菌进行 FISH分析,证实了所分析的多灶性 HCC 样本中存在粪肠球菌和咽峡炎链球菌(图 6A)。通过厌氧和需氧培养成功地从 HCC 患者的新鲜肝匀浆中培养出多种细菌,包括致病菌粪肠球菌。由于这些细菌的丰度与IM-HCC中的转移相关基因相关,他们进一步评估了细菌共培养对HCC细胞侵袭和迁移能力的影响。事实上,伤口愈合实验显示,与对照细胞相比,粪肠球菌(分离自HCC)和咽峡炎链球菌均显著促进伤口愈合(图6B)。Transwell迁移和侵袭实验一致地显示,粪肠球菌和咽峡炎链球菌分别显著增加了HCC细胞的迁移和侵袭(图6C-D)。总之,这些数据表明IM中富集的致病菌粪肠球菌和咽峡炎链球菌可以增强HCC的转移能力。
为了在体内重现这些发现,他们通过将与粪肠球菌、咽峡炎链球菌、大肠杆菌 MG1655(细菌对照)或 PBS(载体)共培养的 RIL-175 肝癌细胞肝内注射到无特定病原体 (SPF) C57BL/6 小鼠体内,建立了原位 HCC 模型(图 6E)。与大肠杆菌或 PBS相比,粪肠球菌和咽峡炎链球菌显著加速了肿瘤生长(图 6E)。Ki-67 染色一致显示,与对照组相比,粪肠球菌和咽峡炎链球菌组的肿瘤细胞增殖显著增加。EMT 标志物的 IHC 分析显示,与对照组相比,粪肠球菌和咽峡炎链球菌治疗组的 E-cadherin 下调,N-cadherin 和 Snail 上调,表明 EMT 升高(图 6F)。值得注意的是,使用抗生素(氨苄西林和庆大霉素)可消除粪肠球菌和咽峡炎链球菌的致瘤作用(图 6E)、增殖作用和 EMT 诱导作用。
随后,他们探讨了致病菌在原位小鼠模型中塑造HCC肿瘤微环境中的作用。通过流式细胞技术对肿瘤内免疫细胞进行了分析。粪肠球菌增加了免疫抑制性髓系来源的抑制细胞 (MDSC) 的比例,尤其是多形核MDSC(图6G)。在粪肠球菌和咽峡炎链球菌治疗组中,都观察到CD8+T细胞和CD4+T细胞减少。此外,CD8+T细胞的效应功能下调,包括粪肠球菌和咽峡炎链球菌中颗粒酶B表达降低,以及咽峡炎链球菌治疗组中IFN-γ表达降低,表明抗肿瘤免疫力减弱(图6H)。总的来说,这些发现表明,IM富集的致病菌粪肠球菌和咽峡炎链球菌可以促进肿瘤生长和转移,并在 HCC 中诱导免疫抑制微环境,突出了这些细菌在调节 HCC 进展和免疫逃避中的关键作用。
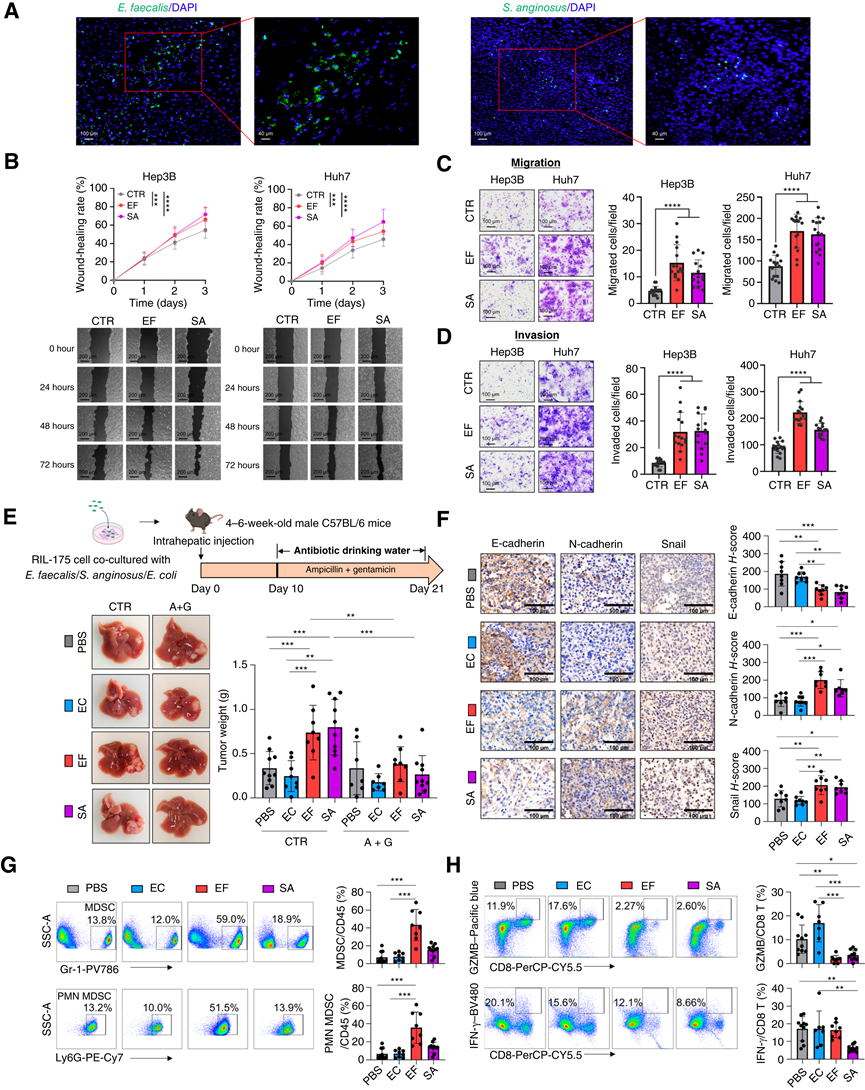
图6. 肝内细菌促进HCC肿瘤生长和转移。
(A) 代表性FISH图像,显示在多灶性HCC患者肝组织中检测到粪肠球菌(左)和咽峡炎链球菌(右)。(B) 伤口愈合试验分析。(C-D) 与 EF 和 SA 共培养的人 HCC 细胞系(Hep3B 和 Huh7)中的迁移和侵袭试验的代表性图像和定量。(E) 说明构建具有原位 HCC 同种异体移植的 SPF C57BL/6 小鼠模型的示意图。(F) IHC 染色代表性图像。(G-H)流式细胞分析。
07
IM富集的细菌从肠道转移到肝脏,促进无菌小鼠罹患肝细胞癌
接下来,他们试图验证IM富集的细菌是否可以从肠道转移到肝脏。无菌小鼠经口灌胃两种IM富集的细菌组合,包括粪肠球菌加咽峡炎链球菌(EF + SA)或粪肠球菌和咽峡炎链球菌加具核梭杆菌(EF + SA + FN),持续 8 周,以大肠杆菌MG1655 或 PBS 作为对照(图 7A)。PCR数据(图 7B)、活菌培养(图 7C)和 FISH(图 7D)数据均证实了相应细菌灌胃小鼠肝脏中存在活菌(粪肠球菌、咽峡炎链球菌和具核梭杆菌),而大肠杆菌和 PBS 灌胃小鼠肝脏检测结果为阴性。
为了确定易位的IM富集细菌是否驱动HCC肿瘤形成,他们通过肝内注射RIL-175(图7E)和Hepa1-6细胞在无菌小鼠中建立了两种原位HCC模型,并用两种IM富集细菌组合(EF + SA或EF + SA + FN)给小鼠灌胃。与RIL-175(图7E)和Hepa1-6模型中的大肠杆菌和PBS组相比,EF + SA或EF + SA + FN均加速了肿瘤生长。肿瘤匀浆中的细菌培养和FISH证实了肿瘤中存在活细菌(图7F)。此外,IM富集的细菌(EF + SA 或 EF + SA + FN)增强了 N-钙粘蛋白和 Snail 的肿瘤表达,而 E-钙粘蛋白下调(图 7G),证实了IM富集的细菌在促进 EMT 中的作用。
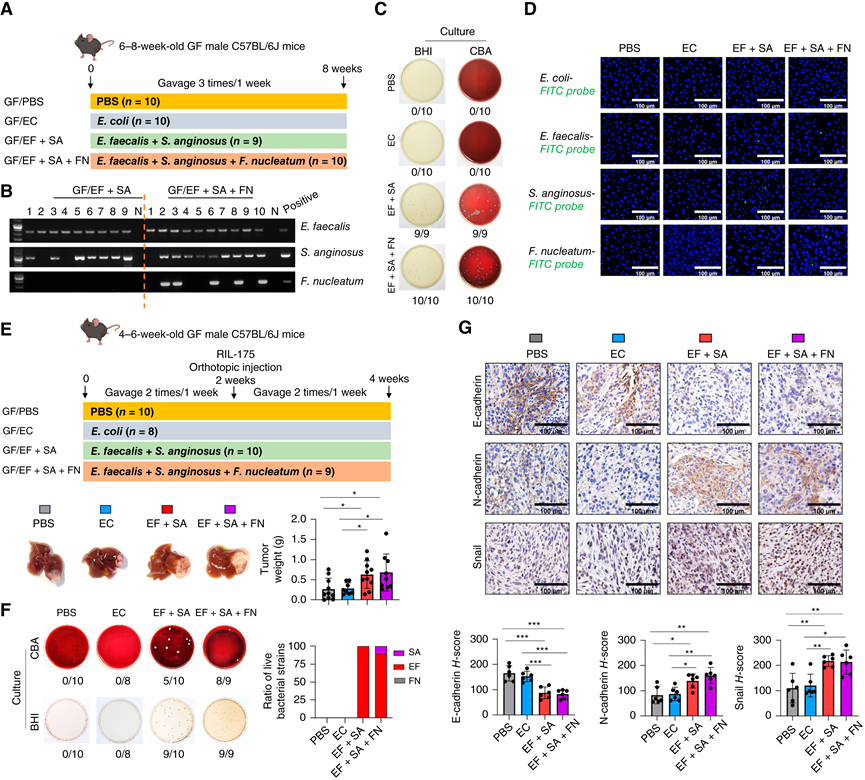
图7. 在无菌小鼠模型中,细菌从肠道转移到肝脏。
(A) 实验设计示意图。(B) 对 GF 小鼠模型肝组织中的细菌 DNA 进行 PCR 分析。(C) 在厌氧条件下从 GF 小鼠肝组织中获得的细菌培养物的图片。(D) 代表性 FISH 图像显示在不同组肝组织中检测到粪肠球菌、咽峡炎链球菌和具核梭杆菌。(E) 显示在 GF C57BL/6J 小鼠中使用 RIL-175 细胞系建立原位 HCC 模型的示意图。(F)厌氧条件下 GF RIL-175 同种异体移植物的细菌培养物的图片。(G) 代表性 IHC 染色图像和定量。
+ + + + + + + + + + +
结 论
本研究对 58 例多灶性 HCC 患者的 242 个 HCC 肿瘤结节和 58 个邻近非肿瘤组织进行了多组学分析,揭示了多灶性 HCC 中的异质性微生物群落。通过革兰氏染色、脂多糖、脂磷壁酸染色和透射电子显微镜证实了 HCC 结节中存在细菌。突变分析将患者分层为IM-HCC 和MO-HCC。IM 和 MO 结节之间的细菌群落存在差异。九种细菌生物标志物组合可以区分 IM 结节和 MO 结节,AUROC 为 0.795。上皮-间质转化通路在IM结节中上调,并与IM富集的细菌相关。IM富集的细菌,例如粪肠球菌和咽峡炎链球菌,通过诱导免疫抑制微环境和上皮-间质转化,促进原位肝癌小鼠模型中的肝癌细胞迁移、侵袭和肿瘤进展。总而言之,肝内微生物群落参与了多灶性肝癌的异质性和发病机制。
+ + + + +



